您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2018-10-17 19:22
CE 认证包括哪 四方面 ?
CE 认证是一个完善的安全保障系统,并非仅仅是将一个样品拿到试验室检验通过而已。
因为 CE 标志是一个安全标志,所以,一个通过CE认证的产品必须确保自产品 的设计,生产,包装,说明书的编写,到运输,销售,产品的整个有效使用寿命 中,以及使用后产品的回收,等等所有环节中,均符合欧洲的健康、安全、与 环境保护之相关法律中所规定的基本要求。因此,一家制造商欲想使其产品通过 CE认证,通常要满足如下4方面的要求:
1.产品投放到欧洲市场前,在产品上加贴CE标签。
2.产品投放到欧洲市场后,技术文件(Technical Files)必须存放于欧盟境 内供监督机构随时检查。
3.对被市场监督机构发现的不合CE要求的产品、或者使用过程中出现事故但是已加贴CE标签的产品,必须采取补救措施。(比如从货架上暂时拿 掉,或从市场中永久地撤除)。
4.已加贴 CE标签之产品型号在投放到欧洲市场后,若遇到欧盟有关的法律更改或变化,其后续生产的同型号产品也必须相应地加以更改或修正,以便符合欧盟新的法律要求。
CE 认证程序
1. 确认出口国家
2. 确认产品类别及欧盟相关产品指令
3. 指定“欧盟授权代表( 欧盟授权代理 ) ”(Authorized Representative)
4. 确认认证所需的模式(Module)
5. 采用 " 自我声明 " 模式还是 " 必须通过第三方认证机构"
6. 建立技术文件 (Technical Files) 及其维护与更新
1、确认出口国家
若出口至欧洲经济区EEA包括欧盟EU及欧洲自由贸易协议EFTA的 30 个成员国 中的任何一国,则可能需要CE认证。
2、确认产品类别及欧盟相关产品指令
若产品属于这里所列22 类中的任何一个,一般地讲,则需要进行 CE认证。若一个产品同时属于一个以上的类别,则必须满足所有类别相对应的产品指令中所列出的要求。
注: 某些产品指令中有时会列出一些排除在指令外的产品。
3、指定“欧盟授权代表( 欧盟授权代理 ) ”(Authorized Representative)
为了能确保前述CE标志 (CE Marking )认证实施过程中的4 项要求得以满足,欧盟法律要求位于30 个 EEA 盟国境外的制造商 必须在欧盟境内指定一家欧盟授 权代表 ( 欧盟授权代理 ) (AuthorizedRepresentative),以确保产品投放到欧洲 市场后,在流通过程及使用期间产品“安全”的一贯性;技术文件 (Technical Files)必须存放于欧盟境内供监督机构随时检查;对被市场监督机构发现的不 合 CE要求的产品、或者使用过程中出现事故但是已加贴CE标签的产品,必须 采取补救措施。(比如从货架上暂时拿掉,或从市场中永久地撤除);已加贴 CE标签之产品型号在投放到欧洲市场后,若遇到欧盟有关的法律更改或变化,其后续生产的同型号产品也必须相应地加以更改或修正, 以便符合欧盟新的法律 要求。
4、确认认证所需的模式(Module)
对于几乎所有的欧盟产品指令来说,指令通常会给制造商提供出几种 CE认证 (Conformity Assessment Procedures)的模式 (Module) ,制造商可根据本身的情况量体裁衣,选择最适合自已的模式。一般地说,CE 认证模式可分为以下9种基本模式 :
1.Module A: internal production control
模式 A: 内部生产控制 ( 自我声明 )
2.Module Aa: intervention of a Notified Body
模式 Aa: 内部生产控制 加第 3 方检测
3.Module B: EC type-examination
模式 B: EC型式试验
4.Module C: conformity to type
模式 C: 符合型式
5.Module D: production quality assurance
模式 D: 生产质量保证
6.Module E: product quality assurance
模式 E: 产品质量保证
7.Module F: product verification
模式 F: 产品验证
8.Module G: unit verification
模式 G: 单元验证
9.Module H: full quality assurance
模式 H: 全面质量保证
基于以上几种基本模式的不同组合,又可能衍生出其它若干种不同的模式。一 般地说,并非任何一种模式均可适用于所有的产品。换言之, 也并非制造商可以随意选取以上任何一种模式来对其产品进行CE认证。
5、采用“自我声明”模式还是“必须通过第三方认证机构”
风险水平 (Risk Level) 较低 (Minimal Risk)
欧盟的产品指令允许某些类别中风险水平 (Risk Level) 较低 (Minimal Risk) 的产品之制造商选择以模式A:“内部生产控制 ( 自我声明 ) ”的方式进行CE 认证。
风险水平较高的产品必须通过第三方认证机构 NB(Notified Body) 介入。
对于风险水平较高的产品,其制造商必须选择模式 A以外的其它模式, 或者模式A外加其它模式来达到 CE 认证。也就是说,必须通过第三方认证机构NB(Notified Body)介入。
模式 A 以外的其它模式的认证过程中,通常均需要至少一家欧盟认可的认证机构 NB 参于认证过程中的一部分或全部。根据不同的模式,NB则可能分别以:来样 检测,抽样检测,工厂审查,年检,不同的质量体系审核,等等方式介入认证过 程,并出具相应的 检测报告,证书等。
目前,已经有 1200 多家认证机构获得欧盟认可, 这些认证机构中的绝大多数位 于欧盟盟国境内。通常情况下, 一家 NB仅被欧盟授权可针对某一类或几类产品 进行某一或几种模式下的认证。换言之,一家欧盟授权的认证机构并不可能针 对所有的产品种类进行认证,即使对其被授权的产品种类,通常情况下也并非 被授权所有的模式。对于每一个欧盟的产品指令,通常都有一个针对该产品指 令的授权认证机构NB名录。
6、建立技术文件 (Technical Files)及其维护与更新
欧盟法律要求,加贴了 CE标签的产品投放到欧洲市场后, 其技术文件 (TechnicalFiles) 必须存放于欧盟境内供监督机构随时检查。技术文件中所包涵的内容若有变化,技术文件也应及时地更新。
"技术文档"是欧盟医疗器械指令中很重要的一个事项, 它的目的是要求企 业准备充份的技术资料和证明, 供主管机关抽查, 或发生诉讼纠纷时使用。 各欧盟指令对于 " 技术档案 " 的要求有所差别, 在这里谨以中国出口企业最 常用的“医疗器械”的要求为例,加以说明。
医疗器械指令 93/42/ EEC要求 " 技术档案 " 可能包含下列项目:
A、企业的质量手册和程序文件
B、企业简介及欧洲代理名称、联系方式
C、CE符合性声明(或称自我保证声明, 若该产品是和其它设备联合运用, 则应有整体符合基本要求的证明材料)
1.产品名称、分类及引用标准条款的简要描述
2.产品概述(包括类型和预期用途)
a) 产品的历史沿革
b) 技术性能参数
c) 产品配合使用的附件、配合件和其它设备清单
d) 产品的图示与样品
e) 产品所用原材料及供应商
3.使用该产品的调和标准 / 或其它标准
4.风险分析评估结论和预防措施( EN1441 产品服务危险分析报告)
5.生产质量控制
a) 产品资料和控制文档(包括产品生产工艺流程图)
b) 产品的灭菌方法和确认的描述
c) 灭菌验证
d) 产品质量控制措施
e) 产品稳定性和效期的描述
6.包装和标识
a) 包装材料说明
b) 标签
c) 使用说明书
7.技术评价
a) 产品检验报告及相关文献
b) 技术概要及权威观点
8.潜在风险评价
a) 产品潜在风险测试报告及相关文献
b) 潜在风险的概要及权威观点
9.临床评价
a) 产品临床测试报告及相关文献
b) 临床使用概述及权威观点
附录 1、产品出厂检测报告
附录 2、产品型式检测报告
附录 3、基本要求检查表
注:
1、临床研究(包括:物理性能,生化、药理 、药动及毒性研究,功 效测试,灭菌合格证明,药物相容性等)
2、生物兼容性测试
( A)EN30993 第一部分要求:细胞毒性、感光性、刺激 - 皮内反应、急性 全身中毒、致热性、亚急性中毒、遗传毒性、植入溶血性;
( B)支持测试:慢性中毒、致癌性、再生性 / 生长性毒素、生物动因退化。)
3、临床资料(需要临床研究或描述临床研究)
4、包装合格证明(EN868)
5、标签、使用说明(EN980、EN1041)
6、结论(设计档案资料的接受、利益对应风险的陈述)
上述文件都必须用欧盟官方语言之一 ( 英、德、法文 ) 编写,但使用说明必须用使用者所在国语言编写。所有文件应在最后一次出货后,至少保存五年。
体外诊断医疗器械 IVDD产品分类
根据 98/79/EC( IVDD)指令附录2 确定产品分类原则对有认证需求的产品进行分类。分类的依据是产品所诊断的疾病。常见产品的分类可参考下表:

与上述诊断试剂配套使用的校准品、仪器、标本采集保存用具均属于体外诊断器 械指令管理的范畴。
医疗器械 MDD产品分类
CE认证过程中判断一个医疗器械正确的分类,仅凭器械的名称是不够的,必须知道完整的预期使用目的(Intended Purpose)!
我们经常听到这样的一句话问题: 某某产品在 CE分类里属于几类医疗器械? 提问者也许不知道仅从一个医疗器械的名称而判断其 CE认证过程中的分类经 常是不妥当的!
1、欧盟与美国的区别 欧盟与美国的医疗器械的分类有很大的不同。
美国的 FDA将医疗器械根据其通用的特点 事先已经分类 并建立了一个公开的数据库可查询;
欧盟则是建立了一套分类规则,让制造商根据产品的预期使用目的(Intended Purpose)按照分类规则自己进行分类。
2、同一个产品,既可以是医疗器械,也可以不是医疗器械
在美国,一个产品是否为医疗器械完全由 FDA决定;
在欧盟,一个产品是否为医疗器械由制造商 ( 申明的产品预期使用目的) 决定 , 比如:电热褥既可以是医疗器械,也可以不是医疗器械。
3、同一个产品,可以是不同类别的医疗器械
比如: 制造商申明的预期使用目的不同, 电热褥既可以是 I 类医疗器械, 也可以是 IIa 或 IIb类医疗器械。
4、同一个产品,作为系统的一部分时与作为配件时属于不同的类别
比如:手术过程中用非主动式抽取腹水装置的留在体外的盛腹水的容器, 作为系统的一部分时可属于IIa 类,但是作为配件时则可属于I 类。
5、类似的产品,可以是不同类别的医疗器械
比如: X光拍片时常用的图像储存通信系统Picture Archiving and Communication Systems (PACS) ,不同制造商申明的预期使用目 ( 功能) 的不同, PACS可以是 I 类医疗器械,也可以是IIa 或 IIb 类医疗器械。
6、类似的产品,有的属于医疗器械 MD, 有的则属于体外诊断器械 IVD
比如:采血管如果 是侵入式的或接触到皮肤的,则属于 MDD 93/42/EEC指令管辖的(普通)医疗器械 MD;
如果 是非侵入式的或完全接触不到皮肤的,则属于IVD 98/79/ec 指令管辖的体外诊断器械IVD。
医疗器械指令 MDD 93/42/eec附录九中详定18 条规则,按医疗产品的危险程度,将产品分为Ⅰ类、Ⅱa类、Ⅱb类、Ⅲ类。
产品分类规则:
1、规则应用由器械的预期使用目的决定;
2、如果器械是和其它器械配合使用,分类规则分别适用于每种器械;
3、附件可以和其它一起使用的器械分开单独分类;
4、启动或影响某种器械的软件与器械属于同一类型。
分类准则:
时间:暂时(<60 分钟)、短期(<30 天)、长期( >30 天)
创伤性:非创伤、通过孔径创伤,外科创伤 、植入。
适用位置:中央循环、中枢神经系统,其它地方。
能量供应:无源,有源。
规则 1~4、所有非创伤性器械均属于I 类,除非他们:
用于储存体液 ( 血袋例外) II a 类
于 Ila类或更高类型的有源医疗器械类II a 类
改变体液成分 II a / II b 类
一些伤口敷料 II a / II b 类
规则 5、侵入人体孔径的医疗器械
暂时使用(牙科压缩材料、检查手套 ) I类
短期使用(导管、隐形眼镜)II a类长期使用(正常牙线) II b类
规则 6-8 、外科创伤性器械
再使用的外科器械(钳子,斧子) I类
暂时或短期使用(缝合针。外科手套) 11a类
长期使用(假关节,眼内晶体 )II b 类
与中央循环系统 (CCS)或中枢神经系统接触的器械 III 类
规则 9、给予或交换能量的治疗器械 II a 类
(肌肉刺激器、电钻、皮肤光疗机、助听器)
一种潜在危险方式工作的II b 类
(婴儿培养箱、高频电刀、超声碎石机、 X 光机)
规则 10、诊断器械
提供能量( 核磁共振,超声诊断仪)IIa 类
诊断/监视体内放射药物分布 II a 类
(r照相机、正电子发射成像仪)
诊断/监视生理功能(心电图、脑电图) II a 类
危险情况下监视生理功能II b 类
(手术中的血气分析仪)
发出电离辐射 (X 射线诊断议) II b 类
规则 11 控制药物或其他物质进出人体的有源器械 II a 类
(吸引设备、供给泵)
如以一种潜在危险方式工作 II b 类
(麻醉机、呼吸机、透析机、高压氧舱)
规则 12 所有其他有源医疗器械属于I 类
(观察灯、牙科椅、轮椅、牙科用治疗灯、记录处理观察诊断图象用的 有源器械)
规则 13、与医用物质结合的器械(含杀精子的避孕套、含抗生素的牙髓材料)III类
规则 14、避孕用具(避孕套、子宫帽II b 类 ) II b/III 类 (子宫内避孕器III 类)
规则 15、清洗或消毒的器械
医疗器械 ( 内窥镜消毒 ) II a 类
接触镜 ( 消毒液、护理液 ) II a 类
规则 16、用于记录X射线图象的器械 (X 光片 ) II a 类
规则 17、利用动物组织的器械(生物)心脏瓣膜、肠线、胶原) III 类
规则 18、血袋 II b 类
各种类型医疗器械的 CE认证步骤
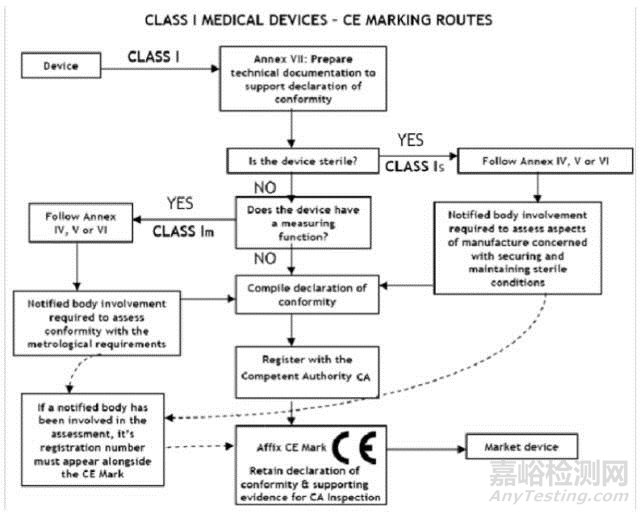
I 类医疗器械的 CE认证步骤
1、分类:确认产品属于I 类医疗器械
2、选择符合性评估途径:请参考下面的流程图
3、编制技术文件
4、CE符合性声明
5、委任欧盟授权代表
6、由欧盟授权代表将制造商及产品在欧盟主管机关注册
7、建立售后警戒系统/ 加贴 CE标签并将产品投放市场
I类医疗器械: CE 符合性评估途径
1、制造商有责任确保其产品符合93/42/eec指令的所有相关的基本要求,必须制定一 份书面(自我)声明来保证。
2、不具备测量功能或非灭菌的 I 类医疗器械(的 CE认证过程中)不需要第三方公告机 构(NB) 参与。 是否符合ISO13485:2003标准,由制造商自愿选择,并非强制性。
3、具有测量功能或灭菌类的 I 类医疗器械(的 CE认证过程中)必须要有第三方公告机 构(NB) 参与。
4、一旦制造商认为其产品符合 93/42/eec 指令的所有相关的基本要求,(欧盟境内的) 制造商,或者(欧盟境外制造商的)欧盟授权代表必须先在欧盟主管机关注册, 然 后才可 加贴 CE标签并将产品投放EEA市场。
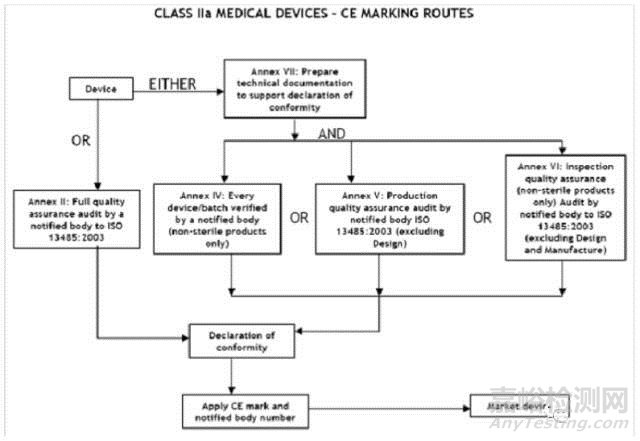
IIa类医疗器械的CE认证步骤
1、分类:确认产品属于IIa类医疗器械
2、选择符合性评估途径:请参考下面的流程图
3、编制技术文件
4、委任欧盟授权代表
5、从第三方公告机构(NB) 获得 CE证书
6、( 完成 )CE 符合性声明
7、将技术文件存放在欧盟授权代表处( 供欧盟主管机关随时检查 )
8、建立 ( 售后 ) 警戒系统/ 加贴CE标签并将产品投放 EEA市场
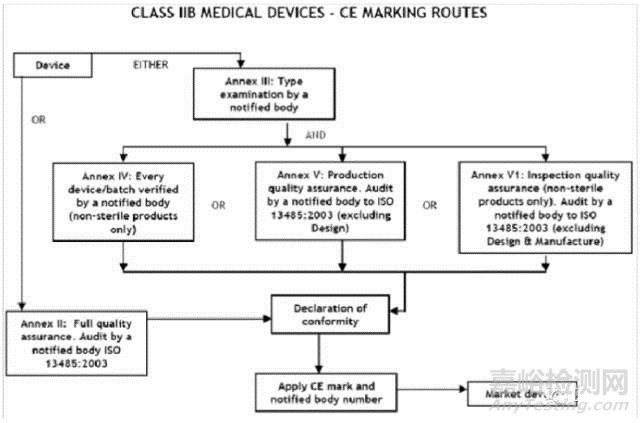
IIb类医疗器械的CE认证步骤
1、分类:确认产品属于IIb 类医疗器械
2、选择符合性评估途径:请参考下面的流程图
3、编制技术文件
4、委任欧盟授权代表
5、从第三方公告机构(NB) 获得 CE证书
6、( 完成 )CE 符合性声明
7、将技术文件存放在欧盟授权代表处( 供欧盟主管机关随时检查 )
8、建立 ( 售后 ) 警戒系统 / 加贴CE标签并将产品投放EEA市场
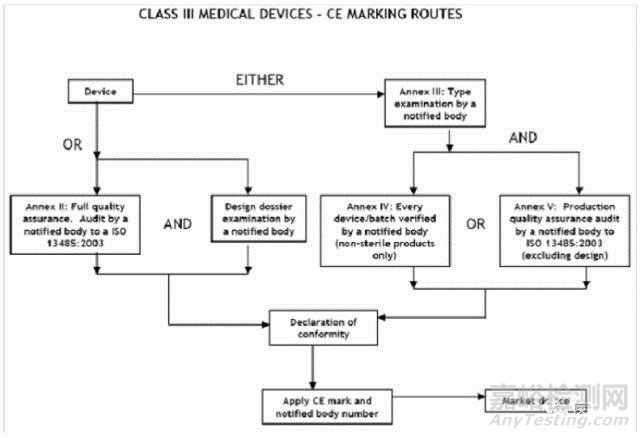
III类医疗器械的CE认证步骤
1、分类:确认产品属于III类医疗器械
2、选择符合性评估途径:请参考下面的流程图
3、编制技术文件
4、委任欧盟授权代表
5、从第三方公告机构(NB) 获得 CE证书
6、( 完成 )CE 符合性声明
7、将技术文件存放在欧盟授权代表处( 供欧盟主管机关随时检查 )
8、建立 ( 售后 ) 警戒系统 / 加贴CE标签并将产品投放EEA市场

来源:AnyTesting


