您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-08-26 17:35
前言
一款仿制药研究时,质量标准研究部分可以参考药典标准,或者是别的法定标准。近年来仿制药的研究思路也已发生了改变,官方机构倡导的仿制药研究也需要科学指导。仿制药是对原研药疗效的仿制,而不是对药物标准的仿制,因此仿制药的标准也可以与原研药不一样。关键在于仿制药是否与原研药药效是否一致,这里通常用生物等效性(Bioequivalency, BE)进行一致性评价,最终确认仿制是否成功。
当我们研究一款新药时,质量研究部分该考虑什么?
质量人员需要与其他药物研发人员协调起来,从药物研发的规律开始,了解药物合成的起始步骤、中间步骤,到最终完成,对药物研发的每一步进行质量研究和制定内控标准。质量标准的建立可以参考药典的规范化过程进行制定。
新药研发质量部分包括质量研究、方法学研究、项目选择、限度和标准制定。
一、质量研究
质量研究包括研制药物对象的特点、制备工艺对药物质量的影响和稳定性影响。
1、原料药质量研究要考虑其结构特征和理化性质;制剂要考虑剂型的特点和临床用途;复方制剂要考虑不同成分之间的相互作用,还有辅料与药物成分之间的相互作用。
常见药物的结构特点如下:
· 镇静与催眠药的苯二氮类含有氮唑结构;
· 抗癫痫药巴比妥类含有丙二酰脲结构;
· 抗精神病药氯丙嗪含有吩噻嗪类结构;
· 镇痛药吗啡含有天然的生物碱结构;
· 哌啶类镇痛药含有4-苯基哌啶结构;
· 解热镇痛药阿司匹林含乙酰水杨酸结构;
· 羧酸类非甾体抗炎药含有的芳基乙酸和芳基丙酸类结构;
· 抗痛风药秋水仙碱为天然生物碱结构;
· 中枢镇咳药可待因中含有的吗啡喃基本结构;
· 平喘药沙丁胺醇中含有的苯乙胺结构;
· 抗溃疡药中西咪替丁含有的碱性芳环和杂原子链结构;雷尼替丁中含有咪唑和氰基胍结构;奥米拉唑含苯并咪唑和亚磺酰基和吡啶环结构;
· 解痉药阿托品中含有的莨菪酸和茛菪醇的结构;
· 胃肠动力药甲氧氯普胺含苯甲酰胺结构;
· 抗心律失常利多卡因含喹啉生物碱结构;普萘洛尔含芳氧丙醇胺结构;抗心绞痛药含硝酸酯类结构;地平类药物含1,4-二氢吡啶类结构;
· 降压类药物卡托普利含巯基和二羧基结构;沙坦类药物含四氮唑结构;
· 降脂类他汀药物含3,5-二羟基羧酸结构;氯贝丁酯含苯氧乙酸类结构;
· 抗肿瘤药物氮芥类烷化剂含b-氯乙胺结构;乙撑亚胺类烷化剂含乙撑亚胺结构;金属配合物含金属铂结构;拓扑异构酶抑制剂羟基喜树碱含有稠和环的内酯生物碱结构;依托泊苷含有鬼臼生物碱结构;多柔比星含有蒽醌类结构;干扰核酸生物合成的抗肿瘤药物含有嘧啶、嘌呤和叶酸类结构。酪氨酸激酶抑制剂替尼类化合物;
每一类药物都有其共性结构,在了解其理化性质的同时,也需要清楚其结构特征。不同的结构特征,会影响后面的分析方法选择,从而影响质量限度的制定。
2、制备工艺对药物的质量影响
原料药质量研究要考虑制备工艺中所用的起始原料、试剂、中间体和副反应产物,以及使用的有机溶剂;制剂通常要考虑所用的辅料、不同工艺的影响,以及可能产生的降解产物。
起始物料的选择影响着原料药的中间体和最终质量标准的制定。因此起始物料的选择要结合原料药自身工艺对杂质的清除效率、结合工艺自身涉及的溶剂、相关的起始物料和遗传毒性杂质(Genotoxic impurity, GTI)而定。起始物料选择一般是原料药母核形成反应的前一、两步的反应物料。过于靠前,质量研究的工作量大;过于靠后,质量标准的要求高。
制剂的工艺通常有固体制剂、液体制剂、半固体制剂和一些现代药物剂剂如靶向制剂、微粒制剂的工艺。常见固体制剂中制粒与混合是关键的步骤,它们会对药物质量产生重要的影响。
3、药物的稳定性
药物在贮藏过程中质量可能发生变化,因此稳定性研究要考察不同条件和包装材料对于药物质量的影响。
稳定性条件一般包括影响因素条件、加速条件和长期条件。影响因素条件一般温度高于加速条件(如60°C),湿度(RH75%或更大),氧化和光照条件。药物强降解实验要关注主要降解杂质,注意降解强度的控制(5%-10%以内),非主要降解杂质,只要不影响主峰的定量可以不关注。
表1:稳定性条件
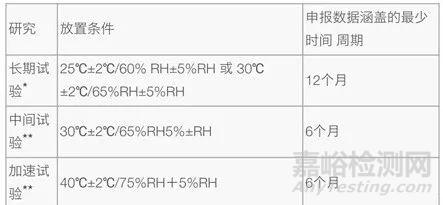
对于稳定性结果的评价应采用系统方法:包括物理、化学、生物和微生物结果。稳定性评价至少要根据三批原料或制剂的结果进行。分析那些可能会随时间变化的定量参数方法是将平均曲线的95%单侧置信限与认可标准的相交点所对应的时间点作为货贺期。降解关系的性质将决定是否可将数据转换为线性回归分析,曲线的拟合用统计方法进行检验。任何评价不仅要考虑含量,还要考虑降解产物和其他相关的性能指标。如有必要,应注意考察药物的质量平衡。
二、方法学研究
分析方法的选择通常基于文献、理论和试验三个方面。常规的检测项目如干燥失重、旋光度、重金属选用药典通用方法,这些方法一般无需验证,直接使用即可。
鉴别项选择方法时要考虑其专属性。针对药物本身,鉴别的方法有化学反应法、色谱法、光谱法、核磁等。化学反应法通常是化合物与某类化学试剂反应显色,不具备专属性。因此鉴别项通常需要选择一项有专属性的色谱法或光谱法。建立鉴别项的分析方法时,包含了两种不同原理的正交方法,对于鉴别才有意义。
检查项要考察方法的专属性、灵敏度和准确性。杂质方法一般使用色谱法,目前常用的方法有外标法,不加校正因子的自身对照法和加校正因子的自身对照法。
选择含量测定方法时,如果杂质方法严格控制,含量测定方法可注重其准确性,可选用容量分析方法。由于UV的专属性差,准确度不及容量法,一般不选用UV方法测定含量。在某些特殊条件情况下,可以选择UV方法进行含量测试,如清洁验证。
有关物质和含量可以采用两种或以上的方法,然后对比择优,选择其中一种方法进行验证,验证项目可参考下表2。
表2:分析方法验证项目
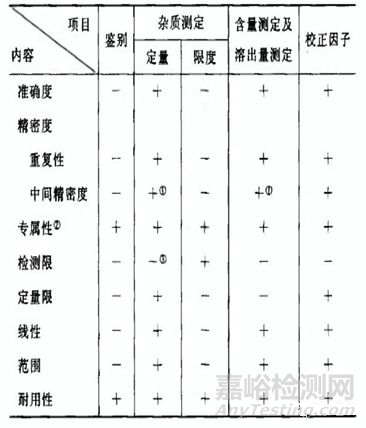
无论是选择非专属性方法,还是专属性方法,分析方法与产品一样,都具有生命周期,每个阶段的目标不一样,分析方法的要求也会不同,关于分析方法生命周期的描述请参看拙作:CMC人士应了解的关于分析方法“生命周期管理”的前世、今生和未来。
三、测试项目
通常的项目选择要基于产品自身的特点,考虑其安全性和有效性,关键的项目一个都不能少。定期或跳检的项目有溶残、微生物和元素分析等。
原药料质量研究的一般内容包括:
•性状:外观,溶解度,熔点,旋光度,吸收系数和密度;
• 鉴别:化学法,色谱和光谱法
• 检查:一般杂质,有关物质,溶残,晶形,粒度,溶液澄清度,干燥失重,水分,异构体。
• 含量测定
制剂质量研究的一般内容包括:
• 性状:样品的外形和颜色;
• 鉴别:化学法和色谱法;
• 检查:含量均匀度,溶出度,释放度,杂质,脆碎度,pH值,异常毒性,残留溶剂
• 含量测定
四、限度制定
1、理化项目的限度
在制定粒径可接受标准时考虑下面四个问题:粒径大小是否影响溶出度、溶解度和生物利用度?粒径大小是影响制剂的生产?粒径大小是否影响制剂稳定性?粒径大小是影响制剂含量均匀度?
如果上面任何一条都受到粒径大小的影响,需要对药物的粒径大小制定可接受标准。
药物的粒度测试一般有激光衍射法和筛分法。制备样品在方法中需要有详细的描述。激光衍射常用D10、D50、D90作为限度控制参数。筛分法通过三个筛号确定累积分布的上下限。激法衍射方法的验证只做精密度和耐用性评价,在方法参数里要注明光学模型,折射率,软件信息。
药物在有多种晶形的情况下,需要开展晶形研究并制定药物晶形参数描述。鉴定晶形的方法有:X-射线-粉末衍射,DSC热分析,显微镜和光谱法。
含不同晶形的药物可能含有不同的溶解度、稳定性和熔点,这些可能会影响制剂的安全性和有效性,因此需要根据这些评估数据,制定药物的多晶形可接受标准。晶形的比例不同,会影响药物制剂的溶出度,安全性或有效性,则在相关的原料药和制剂中制定相应的接受标准。
2、杂质限度
药物杂质分为一般杂质、手性杂质、GTI杂质、重金属杂质、溶剂残留和无机盐。研究时要基于可能存在的杂质,缩小范围,积累杂质的数据。在可能的情况下,基于化学结构、降解途径、合成路线和文献调研进行杂质谱的研究,它是杂质研究的起点。
杂质数量的确定要基于实际工艺,避免因分析方法的原因而漏检某些杂质。工艺研究时,杂质限度设定可以低于前面设定的限度,至少与前面的保持一致,不能高于前面工艺中的杂质限度。
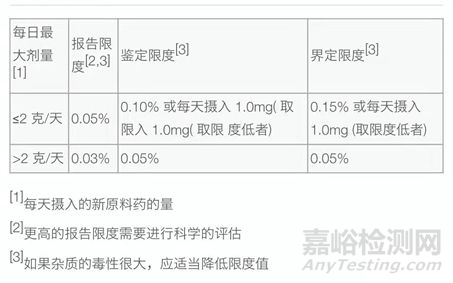
原料药中一般稳定的杂质不需要在制剂中制定。原料药中特定杂质的限度制订时,要积累研发、中试和放大批次的药物杂质数据进行计算,杂质评估结果为测试的平均值和可置信上限(为批分析数据标准差的三倍)的和。如果该杂质不是降解产物,根据表3的规则,依据测试结果判断杂质的限度属于哪一种(报告限、鉴定限或界定限)。
如果杂质为降解产物,要根据加速和长期稳定性研究数据估算药物在复测期内的杂质最大增量,这个增加量与前面计算的杂质平均值的和,同样依据下表规则,进行判断该杂质是否仅为报告,还是需要鉴定,或者质控评估。
表3:原料药杂质限度
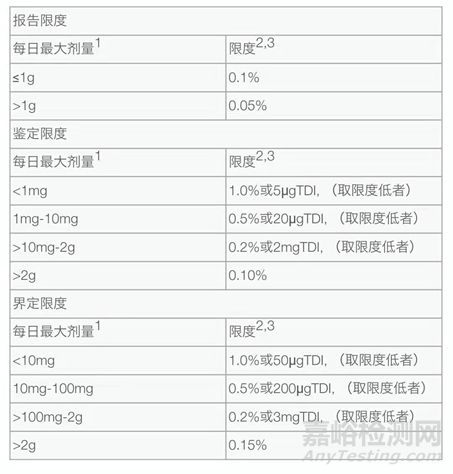
制剂杂质的限度要关注降解杂质,也要关注辅料相容性产生的杂质。制剂中的特定杂质限度制定时,无论杂质是否为储存降解还是生产降解,都需要估算该杂质在降解过程中的最大增量,加上研究批次和生产批次的平均值,依据下表进行判断。
表4:制剂杂质限度表
金属离子铁和铜在某些药物中会起氧化还原反应,影响药物稳定性,因此它们的限度在某些药物中要引起注意。
杂质限度的制定应考虑杂质及含一定限量杂质的药品毒理学研究结果;其他方面包括给药途径,每日剂量,给药人群,杂质药理学研究,原料药来源,治疗周期,在保证用药安全的前提下,企业成本和消费者的成本,等等这些都需要在杂质限度制定时给予考虑。
当药物含有手性碳原子或手性中心时,前面合成步骤需要通过控制原料的手性结构或者某些反应条件,来限制产生手性化合物的反应。当原料药是手性化合物时,制定原料药和制剂的标准时,需要对化合物的手性或对映异构体进行鉴别和手性含量的测定;
有机溶剂限度制定时,我们要了解有机溶剂的分类,主要有三类溶剂,重点关注一类溶剂包括苯、四氯化碳、1,2-二氯乙烷、1,1-二氯乙烯和1,1,1-三氯乙烷,这类溶剂属于遗传毒性杂质,在原料药的合成中要尽量避免。因此在原料的合成中间过程中,如涉及有这类溶剂的残留时,中间体需要对它们进行限量控制,确保最后原料药中这类杂质在可控范围内。
若存在1类或者2类溶剂,应使用专属性的方法,如色谱法,进行定量检查;若只存在3类溶剂,可选用非专属性方法,如干燥失重法,来进行检查。
结束语
药物质量标准分为放行标准和货架期标准。欧盟使用的放行标准和货架期标准通常是一致的,而美、日等国家药物机构常常使用较为严格的放行标准,货架期标准一般要低于放行标准。
首次质量标准中检测方法和接受标准的提出要进行科学的论证,收集研究数据、已有药典标准、毒理临床的数据,以及放大批次和生产验证批次的稳定性数据,分析方法的合理波动等。
质量标准的制定要考虑药物正处于研发的哪个阶段:临床前阶段,还是I期,II期,III 期,或者上市后的IV期。每个阶段对于药物质量标准的要求会有差异,因此可以制定不同的质量标准来适合药物所处的阶段。
临床前研究主要包括药学质量、药效学、药代和安全性评价。药物的有效、安全和可控在这一阶段经过评价后方可进入临床。I期临床的目的是以安全性为主,提供给药方案的依据,为II期提供药剂量的科学依据。II期是疗效评价阶段,探索药物的有效性,为III期提供科学基础。III期临床是药物疗效确证阶段,为扩大的多中心试验。
药物的每个研究阶段对于安全性和有效性都将进行探索,每个阶段有着不同的研究目的和要求。因此测试项目和分析方法的选择也是一个逐步确定下来的过程,药物的质量标准也是一个逐步评价和探索的过程。
参考文献
1.CDE,2005《化学药物杂质研究的技术指导原则》
2.CDE,2005《化学药物质量标准建立的规范化过程技术指导原则》
3.CDE,2005《化学药物质量控制分析方法验证技术指导原则》
4.ChP 2015: <9101 药品质量标准分析方法验证指导原则》
5.ICHQ1A: StabilityTesting of New Drug Substances and Products
6.ICHQ3A: Impuriteisin Drug Substances
7.ICHQ3B: Impurties inNew Drug Proucts
8.ICHQ3C: Impurities:Guideline for Residual Solvents
9.ICHQ6: Specifications:Test Procedures and Acceptance Criteria for New Drug Substances and New DrugProducts: Chemical Substances

来源:曾文亮、药事纵横


