您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-03-11 10:13
在《中国药典》2020年版四部通则增修订内容(第七批)0107栓剂(2020 版第一次征求意见稿)项下规定:栓剂系指原料药物与适宜基质等制成供腔道给药的固体制剂。栓剂因施用腔道的不同,分为直肠栓、阴道栓和尿道栓。直肠栓为鱼雷形、圆锥形或圆柱形等;阴道栓为棒状。阴道栓可分为普通栓和膨胀栓。阴道膨胀栓系指含药基质中插入具有吸水膨胀功能的内芯后制成的栓剂;膨胀内芯系以脱脂棉或粘胶纤维等经加工、灭菌制成。栓剂作为一种非口服局部给药的固体制剂,克服了口服制剂中一些难以避免的不足,如药物不受或少受胃肠道pH值或酶的破坏,避免药物对胃黏膜的刺激性,避免肝脏首过效应,适宜不便口服给药的患者等。近年来该剂型发展迅速,相继出现了中空栓剂、双层栓剂、泡腾栓剂、海绵栓剂、凝胶栓剂、微囊栓剂、渗透泵栓剂、缓释栓剂以及不溶性栓剂等新型栓剂。随着新型栓剂的增加以及科学研究的不断深入,药典中收载的评价方法难以对栓剂进行全面的质量评价,本文对当前的研究进展和法规要求进行了简要整理分析,旨在为栓剂质量研究者提供一些思路和参考。
一、相关指导原则
中国药典2015年版四部通则0107栓剂
中国药典2020年版四部通则增修订内容(第七批)0107栓剂(2020 版第一次征求意见稿)
中国药典2015年版四部通则0922融变时限检査法
国家食品药品监督管理总局药品审评中心《局部作用阴道制剂(阴道片、阴道栓)仿制药的评价技术要求(征求意见稿)》20181024
欧洲药典2.9.22 Softening time determination of lipophilic suppositories
二、质量研究内容
在仿制药研究中,根据目标产品的质量概况(QTPP)确立制剂的关键质量属性(CQA),阴道栓的CQA包括但不限于以下研究:性状(如形状、尺寸、硬度、重量、体积等)、融变时限(融变过程)、pH值(或酸度)、溶出度(或释放度)、有关物质、含量测定、微生物限度、抑菌剂及抗氧剂含量、原料的晶型和粒度等。以及模拟阴道制剂体内释放行为的体外释放试验对比研究资料。
性状
栓剂的性状可能影响药物在腔道内的分布,从而影响药物的释放行为,一般认为,仿制品的性状(如形状、尺寸、硬度、体积、重量等)应与参比制剂一致。栓剂外观应完整光滑,颜色均匀一致,无软化、变形或干裂,药物和基质混合完全。
鉴别
标准中的常规项目,如建立HPLC法、UV法等进行鉴别。
pH值
目前2015年版中国药典中未规定栓剂进行pH值检查,而《局部作用阴道制剂(阴道片、阴道栓)仿制药的评价技术要求(征求意见稿)》,要求新注册分类4及5.2类的局部作用、局部起效的阴道制剂(阴道片、阴道栓)应关注栓剂的pH值,应与参比制剂一致。对于油溶性基质的栓剂可不要求进行pH值考察。这可能是因为正常健康妇女阴道pH值在4.0~5.0之间,若药物改变了阴道酸碱度,长期使用将对正常菌群产生影响,使条件致病菌增殖而生产疾病,出于阴道用制剂的酸碱性引起的安全性问题考虑,而在阴道给药制剂中增加pH值检查研究。
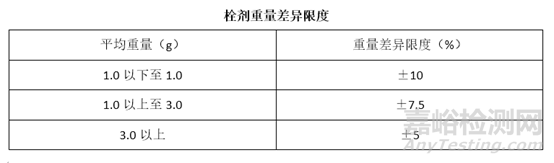
重量差异
取供试品10粒,精密称定总重量,求得平均粒重后,再分别精密称定各粒的重量。每粒重量与平均粒重相比较(有标示粒重的中药栓剂,每粒重量应与标示粒重比较),按表中的规定,超出重量差异限度的不得多于1粒,并不得超出限度1倍。凡规定检查含量均匀度的栓剂,一般不再进行重量差异检查。
融变时限
《中国药典》2015版四部规定对栓剂进行的融变时限检查,方法为通则0922融变时限检査法。规定脂肪性基质的栓剂3粒均应在30分钟内全部融化、软化或触压时无硬心;水溶性基质的栓剂3粒在60分钟内全部溶解。如有1粒不符合规定,应另取3粒复试均应符合规定。栓剂的释药速度与融变时限有关,应对仿制品和参比制剂的融变时限(包括融变过程)进行比较研究。
膨胀值
在《中国药典》2020年版四部通则增修订内容(第七批)0107栓剂(2020 版第一次征求意见稿)中新增了膨胀值检查法,规定:除另有规定外,阴道膨胀栓应检查膨胀值,并符合规定。检查法取本品3粒,用游标卡尺测其尾部棉条直径,滚动约90°再测一次,每粒测两次,求出每粒测定的2次平均值(Ri);将上述3粒栓用于融变时限测定结束后,立即取出剩余棉条,待水断滴,均轻置于玻璃板上,用游标卡尺测定每个棉条的两端以及中间三个部位,滚动约90°后再测定三个部位,每个棉条共获得六个数据,求出测定的6次平均值(ri),计算每粒的膨胀值(Pi),三粒栓的膨胀值均应大于1.5。Pi=ri/Ri
软化时间
欧洲药典通则2.9.22 Softening time determination of lipophilic suppositories中规定进行栓剂的软化时间考察,本项目是欧洲药典通则要求的必检项目,能更好地模拟栓剂在体内的软化时间,一般在36±0.5℃条件下,样品的软化时间应小于20min。应对仿制品和参比制剂的软化时间进行比较研究。
溶出度(释放度)
溶出度系指活性药物成分在规定条件下从制剂中溶出的速率和程度,它是评价和控制药物制剂质量的重要指标,对评估制剂的批次质量、优化处方及制备工艺、保证处方工艺等变更前后产品质量的一致性有重要作用。尽管国内外药典栓剂标准项下大多数未列溶出度检查项,《中国药典》中也未收载栓剂的溶出度/释放度方法,未规定进行溶出度/释放度检查项,但《局部作用阴道制剂(阴道片、阴道栓)仿制药的评价技术要求(征求意见稿)》,要求新注册分类4及5.2类的局部作用、局部起效的阴道制剂(阴道片、阴道栓)应参照溶出度相关技术指导原则对仿制药和参比制剂进行系统的体外释放对比研究。因此溶出是必须要研究的内容。欧洲药典规定栓剂采用流池法测定溶出,该法在2015版中国药典中尚未收载,但2020版中国药典中已收载,因此,以后研究栓剂的溶出使用流池法也符合药典要求了。如果企业没有流池法装置,试验中也可采用普通溶出仪(如篮法或桨法,或其他可行的改良装置),栓剂样品可采用自制沉降圈置于杯底,于不同时间点取出样品测定,绘制溶出曲线。由于正常健康妇女阴道pH值在4.0~5.0之间,研发中可以根据常释或缓释栓剂等特点,选择此范围内或其他溶液(包括水)为介质进行研究(必要时加入适量表面活性剂,如SDS),转速可为25~100rpm(模拟阴道环境宜选择较慢的转速),体积常用500~1000ml或更少。
体外释放试验
建议结合阴道生理环境和临床用药特点(如阴道pH、给药环境的液体量及药物在阴道内存留的时间等)进行模拟阴道制剂体内释放行为的体外释放研究,如采用人工模拟阴道液或模拟阴道pH的弱酸性介质、采用模拟阴道制剂体内释放的溶出装置,兼顾临床用药方法确定释放试验考察时间等,应提供体外释放试验系统的研究及验证资料,并在此基础上,对比仿制药与参比制剂的释放行为。申请人可根据产品特点,设计与临床局部作用相关的体外释放一致性研究,以佐证药物与参比制剂的临床疗效一致性。朱秀城等采用自制的体外黏附时间测定装置和Setnikar-Fantelli 装置模拟阴道环境,测定了栓剂的黏附时间、液化时间和药物滞留量;采用转篮法和HPLC法测定黄体酮生物黏附缓释栓中药物的体外释放度。此文可以作为研究时参考。
体内吸收试验
可选择家兔为试验动物,开始时剂量不超过口服剂量,以后再两倍或三倍地增加剂量。给药后按一定时间间隔抽取血液或收集尿液,测定药物浓度。最后计算动物体内药物吸收的动力学参数和AUC等。
有关物质
应根据产品的质量特点,按照相关技术指导原则以及国内外药典的收载情况,科学合理的选择有关物质的检查方法,并提供规范的方法学验证资料。并按相关技术指导原则的要求,参考参比制剂的实测结果和国内外药典收载的杂质指标,制定合理的有关物质限度。降解产物:重点对制剂的降解产物进行研究,包括原料药的降解产物或者原料药与辅料和/或内包材的反应产物,分析原料药降解途径。原料药的工艺杂质一般不需在制剂中进行监测或说明。异构体:对于存在几何异构体和手性异构体等情况,应根据产品特点和生产工艺等方面的研究,确定是否订入标准。遗传毒性杂质:应结合药物的吸收途径,根据相关文献、参比制剂的情况,通过对生产工艺、产品降解途径的分析,判断是否可能产生潜在的遗传毒性杂质,必要时进行针对性研究,根据研究结果按照相关技术指导原则进行控制。
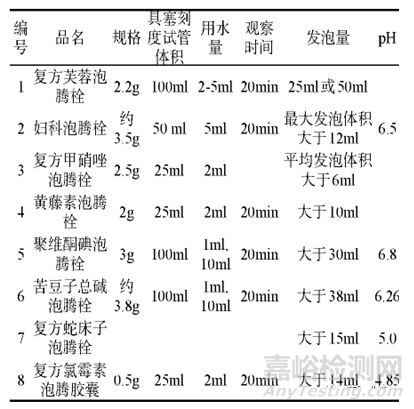
发泡量
泡腾栓剂是在栓剂中加入碳酸氢钠等于有机酸组成的泡腾剂,在使用过程中,栓剂与腔道液体接触时,泡腾剂发挥作用,与泡腾片机制相似,可加速栓剂的熔融和药物释放。泡腾栓剂在研究中,应增加发泡量检查,但限度设置需要有合适的理由。几种泡腾栓剂的发泡量检查条件举例如下:
含量测定(含量均匀度)
对于主成分,含量和含量均匀度均应符合规定,其中含量均匀度应符合药典通则0941含量均匀度检查法项下规定。对于抗氧剂、抑菌剂等成分,应建立方法进行考察,并制定限度。
刺激性试验
由于栓剂为经黏膜给药。因此考察栓剂对黏膜的刺激性有助于时栓剂进行安全性评价,黏膜刺激性检查一般是采用动物试验进行,即将基质检品的粉末、溶液或栓剂,施于家兔的眼黏膜上,或纳入动物的直肠、阴道,观察有何异常反应。在动物试验基础上,临床验证多在人体肛门或阴道中观察用药部位有无灼痛刺激以及不适感觉等反应。
微生物限度
中国药典通则规定,除另有规定外,照非无菌产品微生物限度检査:微生物计数法(通则105)和控制菌检查法(通则1106)及非无菌药品微生物限度标准检查,应符合规定。
三、稳定性研究内容
稳定性研究一般包括影响因素试验、加速试验和长期试验,必要时应考察中间条件下的稳定性。对于内包材无法起到完全遮光保护的品种,应按照《化学药物(原料药和制剂)稳定性研究技术指导原则》的要求进行光照稳定性研究。
中国药典通则9001《原料药物与制剂稳定性试验指导原则》原料药物及制剂稳定性重点考察项目参考表中规定栓剂应进行性状、含量、融变时限、有关物质的检查。但是目前稳定性中仅进行这些考察还是不够的。
根据《局部作用阴道制剂(阴道片、阴道栓)仿制药的评价技术要求(征求意见稿)》栓剂的性状可能影响药物在腔道内的分布,从而影响药物的释放行为,建议在稳定性性状项下增加形状变化、硬度等考察,并提供支持运输条件短暂偏离贮藏条件的其他稳定性数据(如低温或冻融试验),如:栓剂基质在贮存过程中可能发生因受热而黏着、熔化,造成变形,在运输和贮藏过程中因撞击而破碎,导致药物吸收发生改变。
另外,在稳定性末期应与参比制剂进行释放度对比研究;如处方中含有抗氧剂、抑菌剂等辅料,在稳定性研究中还应定量地考察这些辅料的变化情况。有的品种必要时还需要考察水分含量。
四、总结
1、溶出度和体外释放试验已成为栓剂的重要质量评价指标,溶出装置有篮法、桨法、流池法,方法也可以在药典方法基础上进行改良。
2、仿制栓剂应与参比制剂对比pH、软化时间、溶出、膨胀值、发泡量等易忽视的指标。
3、2020版药典相关通则应该进行必要完善或修订,如与审评技术指导原则、国外药典同剂型评价方法统一。
参考文献
1、潘卫三.工业药剂学[M].北京:中国医药科技出版社,2015.8
2、王雨青,訾鹏,刘瑾.硝基咪唑类栓剂的研究[J]. 中国医药工业杂志,2019-07-24
3、朱秀城,张蜀,邓红等.黄体酮生物黏附缓释栓的体外评价方法研究[J].广东药科大学学报,2017,33(2):148-152
4、孙学志,杨杨,刘利群等.《中国药典》附录栓剂的修订设想[J].黑龙江医药,2015,28(3):510-512

来源:梅希/药事纵横


