您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2020-03-19 10:52
随着新型冠状病毒SARS-CoV-2*(以下简称“新型冠状病毒”)引起的疫情在全球范围内传播扩散,在疫情防控中,很多国家/地区包括欧盟都面临检测试剂紧缺的状况。
新型冠状病毒检测试剂出口到欧盟,应该满足什么法规要求?7个常见问答,帮助制造商看懂欧盟法规要求。
*注:对于新型冠状病毒,WHO曾称其为“2019-nCoV ”,但后来确定的官方正式名称为“SARS-CoV-2”,目前国内仍多采用“2019-nCoV”,本文为了保持与国际上对该病毒名称的一致,故采用“SARS-CoV-2”。
01、新型冠状病毒检测试剂须满足哪个法规要求?
新型冠状病毒检测试剂进入欧盟市场需要进行CE认证。目前正处在从IVDD指令(98/79/EC)到IVDR法规(Regulation (EU) 2017/746)的过渡期,新型冠状病毒检测试剂制造商既可以选择遵照IVDD指令的要求,也可以选择遵照IVDR法规的要求,通过CE认证,合法上市。
02、新型冠状病毒检测试剂在IVDD及IVDR之下的分类各是怎么样的?认证途径的主要区别是什么?该选择哪个法规下认证途径比较合适?
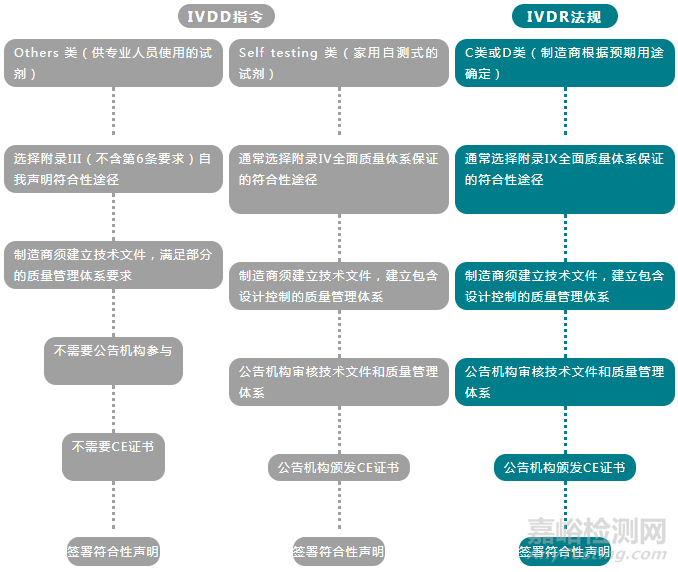
注:鉴于目前市场上绝大多数新型冠状病毒检测试剂都是供专业人员使用,在IVDD下分为Others 类的产品。所以以下问题/建议仅是针对Others 类产品,不适用Self testing 类产品。
因IVDR下的CE认证需要公告机构审核,耗时较长,而且目前IVDR下,只有少数公告机构获得资格,业务尚未完全开展。鉴于目前疫情严重,制造商可优先选择IVDD下的自我声明的途径,进入欧盟市场,这样既可以符合法规要求,又可以快速将产品投放市场,后续在过渡期结束(2022年5月26日)之前,再按照IVDR法规要求,完成CE认证。
03、如果选择IVDD的自我声明符合性途径,有什么特别注意的事项?
即使按照自我声明符合性途径的要求,制造商也同样需要建立质量管理体系、建立技术文件、进行售后监督,只不过无须公告机构来进行审核。此外,对欧盟以外的制造商(比如中国的制造商)还须指定欧盟代表,并在主管当局注册,产品才能够上市销售。
04、对质量管理体系有什么要求,是否一定需要符合ISO 13485中所有要求?
ISO 13485是欧盟在质量管理体系方面的协调标准,自我声明途径下的质量管理体系,只要求在生产、检验、管理、售后监督方面进行控制,并不需要覆盖 ISO 13485的所有要求(比如设计可以删减),也不需要通过ISO 13485认证即可满足法规要求。但是在实际操作过程中,按照ISO 13485的适用要求建立质量管理体系并进行认证,可以提高客户对制造商能满足法规要求及产品质量保证的信心。
05、IVDD下的技术文件应该包含什么内容?
技术文件主要包括:
所认证的产品及各种规格型号的描述
各种组分、设计原理、技术性能、生产工艺等描述
如含人源性物质,对其来源的说明
风险管理报告,基本要求检查表及对应的协调标准
测试报告
性能评估报告
标签及说明书
稳定性报告等
具体内容请参照IVDD 98/79 EC指令附录III第三章节。技术文件是产品符合IVDD法规要求的证据集合,是在CE认证中需准备的最重要的文件,其内容和作用类似于在中国注册时,向药监局提供的注册文件。技术文件还须提交给欧盟代表,以备主管当局可能的检查。
06、是否一定需要欧盟代表,欧盟代表要做哪些工作?
在IVDD之下,对于非欧盟境内的制造商,需要有欧盟的授权代表,欧盟代表是制造商与主管当局沟通的桥梁,承担着保存技术文件、不良事件报告、注册等职责。制造商须与欧盟代表签订书面协议,对这些职责加以规定。产品在主管当局的注册可由欧盟代表完成。欧盟代表的地址信息也需要在产品的包装上体现。
07、新型冠状病毒检测试剂的性能须满足什么要求?
目前, 欧盟尚未对新型冠状病毒检测试剂的性能评估要求给出具体的指导性文件,但
FDA 发布了《emergency use authorization (EUA) interactive review template for molecular-based single target tests for sars-cov-2 that causes coronavirus disease 2019 (covid-19)》
WHO发布了《Instructions for Submission Requirements: In vitro diagnostics (IVDs) Detecting SARS-CoV-2 Nucleic Acid》
这两份文件中包含了FDA和WHO在各自的应急审批程序下,对新型冠状病毒核酸检测试剂在分析性能,临床性能方面的要求,这些要求可以看作是欧盟法规中所要求的 “state of the art” ,有重要的参考价值,制造商可以参照这些文件进行性能评估。
除此之外,中国发布了:
《2019新型冠状病毒核酸检测试剂注册技术审评要点》
《2019新型冠状病毒抗原-抗体检测试剂注册技术审评要点》
其中多数要求与FDA、WHO的要求类似,但也有不同之处。因为语言和文化方面的差异,欧盟更了解FDA和WHO的一些要求,所以如果有差异,请参照FDA或WHO的要求,或者加入适当的理由说明。

来源:BSI曾强松


