您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-09-16 18:05
关于专属性,中国药典给出的定义是:在其他成分存在的条件下,采用的分析方法能够正确测出被测物的能力。鉴别反应、杂质检查和含量测定方法均应考察专属性。药典中就鉴别反应、杂质检查和含量测定的专属性均做出了详细说明。在实际验证过程中,大多数情况下我们会按部就班去做。但有一些方法,其属于非专属性方法,如滴定法,其专属性只可以通过其他方法去佐证。本文就专属性的实际操作及相关问题进行简单论述,不足之处还请指正。
一、专属性的具体做法讨论
本文主要就杂质检查和含量测定方法的专属性做法进行论述。
1、含量测定
药典描述:对于含量测定,在杂质对照品可获得的情况下,可在试样中加入杂质或辅料,考察测定结果是否受干扰,并与未加入杂质或辅料的试样比较测定结果。在杂质或降解产物不能获得的情况下可用另一个经验证了的方法或药典方法比较结果,也可用强光照射、高温、高湿、酸(碱)水解或氧化等方法进行加速破坏,以研究可能存在的降解产物对含量测定的影响。含量测定应对比两种方法的结果。
在药典和ICH中,含量测定与杂质检查的专属性没有明确的分开描述,所以实际中大部分情况下含量测定与杂质检查的专属性做法基本保持一致。这里有以下几点需要考虑:
(1)含量测定方法与杂质检查方法完全相同。
在此情况下,既需要考察杂质能够准确测定,又需要考察含量测定的准确性。由于供试品浓度完全一致,在杂质和辅料存在的情况下,需要着重考察杂质和辅料与主成分的分离情况以及杂质与辅料、杂质与杂质间的分离情况,对于含量测定,一般要求杂质和辅料均能与主成分有效分离。其次是进行破坏试验,一般要求有一种或几种破坏能够得到相应的降解产物,观察降解产物与主成分峰的分离度,并进行峰纯度检查。在含量测定方法与杂质检查方法完全相同的情况下,以上做法是最常见的,但这种做法忽略了考察含量测定本身的准确性,仅考察了主成分不受干扰的情况,忽略了测定的重点的含量。
(2)含量测定与杂质检查方法色谱条件相同,但供试品浓度不同。
在此种条件下,往往是含量测定供试品浓度远低于杂质检查的供试品浓度。首要考虑的是,当供试品浓度较低时,杂质和辅料在该色谱条件下是否会出峰。具体的做法:使用与主成分相邻杂质对照品按照限度的120%或150%配制相应浓度的溶液,在该色谱条件下进样,观察出峰情况。按照处方量配制相应的辅料溶液,在该色谱条件下进样,观察出峰情况。这里涉及到含量测定供试品浓度选择的问题,一般会选择限度的120%或150%的杂质不出峰对应的浓度作为含量测定供试品浓度。在此种情况下,考察主成分与杂质或辅料的分离情况意义不大,降解试验也不适用。
(3)含量测定与杂质检查方法完全不同。此种条件下具体实施方法需要根据供试品浓度的选择情况而定。
如以上所述,三种情况下均需要考虑含量测定的准确性。就外标法测定含量来讲,需要考察在杂质和辅料存在的情况下含量测定的结果不受影响。具体的操作如下:
按照含量测定供试品浓度,使用原料与限度浓度的相邻杂质配制成溶液1;
按照含量测定供试品浓度,使用原料与处方量辅料配制成溶液2;
向供试品中加入限度浓度的杂质,配制成溶液3;
供试品溶液作为对照溶液。
分别取溶液1、2、3和对照溶液测定含量,对比含量测定结果,要求结果应无明显差异。取供试品进行适量破坏试验,配制成相应的供试品溶液,进行含量测定,测定结果与未破坏含量结果应无明显差异。这里所说的适量破坏试验,降解产生1%~2%的杂质即可,破坏程度较强,此种对比就会失去原本的意义。
2、杂质检查
药典描述:在杂质对照品可获得的情况下,对于杂质检查,可向试样中加入一定量的杂质,考察各成分包括杂质之间能否得到分离。在杂质或降解产物无法获得的情况下,可用另一个经验证了的方法或药典方法比较结果,也可用强光照射、高温、高湿、酸(碱)水解或氧化等方法进行加速破坏,杂质检查应对比检出的杂质个数,必要时可采用光二极管列阵检测器和质谱检测进行峰纯度检查。
对于杂质检查的专属性,一般做法:按照杂质检查供试品浓度,使用原料和限度浓度的杂质配制成混合溶液;取各杂质配制成各单个杂质溶液;按照处方量取辅料混合制成辅料溶液;分别取空白溶剂、混合溶液、各单个杂质溶液和辅料溶液进样,进行相应峰的定位,以说明各杂质峰、辅料峰、空白溶剂峰和主成分峰的分离情况。取供试品进行破坏试验,观察降解产物与主成分峰的分离情况,使用光二极管列阵检测器进行峰纯度检查。个人认为在应当使用光二极管列阵检测器和质谱检测两种方法同时进行峰纯度检查。
二、专属性相关的一些问题
1、杂质检查专属性中破坏试验的破坏程度
关于破坏试验的破坏程度,目前较为广泛认可的是5%~10%。个人认为尚不能如此定论,具体从以下几点进行论述:
(1)需要考虑方法建立之初杂质能够分离的承载量,即杂质与杂质之间、主峰与相邻杂质之间能够有效分离时的最大浓度。
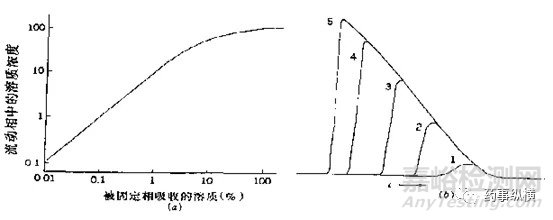
图1 样品量对峰宽与峰形的影响

图2(上)、3(下)杂质与杂质之间、主峰与杂质之间的分离情况
如图1,列出了样品量对峰宽和峰形影响的一种情况。当溶液中样品量增加时,峰宽会相应变宽。如图2(上)所示,杂质2与杂质3在相应限度的情况下可以达到基本分离,但若破坏试验时杂质2与杂质3的量超过限度,则会因峰宽变宽影响二者之间的分离度。如图3(下)所示,杂质Y与主峰1之间完全分离,但若破坏试验时杂质Y的量超过限度很多(如杂质Y达到0.5%),则会由于其峰宽变宽影响与主峰的分离度,导致主峰峰纯度达不到要求。
(2)药物稳定性本身的考虑。
对药物进行加速破坏试验,一是为了了解药物的降解途径及相应的降解产物,二是为了考察降解产物在该分析方法下能不能被准确检出。一般情况下,光照破坏采用可见光及近紫外条件下室温放置1~3天,酸碱破坏最高使用1mol/L强酸强碱室温条件放置1天,氧化破坏最高使用30%过氧化氢溶液室温放置1 天,高温可采用60~80℃最长放置5天,便可知晓药物在相应条件下的稳定性;选用更加苛刻的条件进行破坏并没有多大意义,需要结合实际中遇到的情况。
(3)、与物料平衡之间的相互关系。
计算物料平衡,是为了判断使用该检测方法能否将产生的降解杂质全部准确检出,破坏程度较强会直接影响物料平衡。
2、物料平衡的可接受范围
目前所接触到的一些资料中,物料平衡的可接受范围基本定为90%~110%,但这在药典中并没有明确的规定。个人认为需要就实际情况而论。影响物料平衡的因素有以下几个:
(1)杂质的响应因子
在进行分析方法验证时,我们需要得到杂质的相对校正因子,一般会存在有杂质相对校正因子为0.9以下或1.1以上的,表明杂质在方法中的响应差异较大,同样也会存在未知杂质响应差异较大的情况。当进行加速破坏试验后,由于响应差异较大的原因,杂质的峰面积不能与主峰失去的峰面积划等号,因此物料平衡算出来就不是100%。
(2)有未检出的杂质
当破坏程度较为剧烈时,有可能会产生方法检不出的杂质,其可能为生成的气体、无机盐、在该条件下没有响应的物质、由于检测时间较短在规定时间内无法出峰的物质等。由于破坏后失去了该部分物质,导致物料平衡算出来不是100%。
(3)杂质的范围
在相应的浓度范围内,浓度与响应值呈线性关系(或其他非线性模型),当超出该范围,响应值会明显不符合该种关系。若破坏程度较为剧烈,有杂质的响应值超出了其相应范围内的某种特定关系,也会导致物料平衡算出来不是100%。
以上是列出的其中三种可能存在的因素,也可能存在其他因素。
验证中的项目相互之间均有联系,如在归纳物料平衡计算时结果不是100%的原因时,杂质的校正因子和线性范围便可以进行相应的解释。

来源:药事纵横


