您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-11-28 17:42
一、前言
遗传毒性杂质(Genotoxic Impurities,GTIs),也称基因毒性杂质,是指能引起遗传毒性的杂质,包括致突变杂质和其他类型的非致突变杂质。其中,致突变杂质(Mutagenic Impurities)指在较低水平时也能直接引起DNA损伤,导致DNA突变,从而可能引发癌症的遗传毒性杂质[1],其致突变致癌作用机制目前一般认为是线性机制。致突变性(Mutagenicity)通常由标准的细菌回复突变试验(Ames试验)结果判定,致癌性(Carcinogenicity)由动物致癌试验结果和人类致癌性相关证据判定。非致突变性遗传毒性杂质不直接作用于DNA,一般引起染色体畸变,通常为阈值机制,即化合物剂量与效应之间的关系不完全是线性的,在浓度达到一定阈值后才产生相应的毒性作用。阈值机制的存在可能是化合物在与DNA接触前即被降解失活,或产生的损伤一定程度内可被有效修复[2]。随着科学研究的深入,逐步发现阈值机制在非致突变(如苯胺)和致突变(如甲磺酸乙酯EMS)致癌物中都存在。阈值机制在《Genotoxic Impurities:Strategies for Identification and Control》[3]一书第7章节有详细阐述。ICH M7和中国药典2020版四部通则9306均关注致突变机制的遗传毒性杂质,非致突变机制的遗传毒性杂质以一般杂质水平存在时,通常可忽略其致癌风险。
二、法规发展历程
自2000年起,药监机构相继发布相关文章或指导原则,要求原料药和制剂厂商对遗传毒性杂质进行风险控制。如下为主要的法规发展历程:
2000年,欧洲药典公开发表文章,提出磺酸在醇溶液中成盐可能产生磺酸酯类杂质;
2002年,CPMP(Committee for Proprietary Medicinal Products, 欧洲专利药品委员会),现为CHMP(Committee for Human Medicinal Products,人用药品委员会),发布《Position paper on the limits of GIs》,提出阈值机制和线性机制,需要对遗传毒性杂质进行质量(Quality)和安全性(Safety)评估;
2004年, 《Guideline on the Limits of GIs-draft》发布,提出ALARP(As Low As Reasonably Practical,合理可行的最低程度)原则和TTC (Threshold of Toxicological Concern,毒理学关注阈值)概念;
2006年,PhRMA(Pharmaceutical Research and Manufacturers of America,美国药品研究和制造商协会)发布白皮书,提出分期TTC(Stage TTC)概念,并将遗传毒性杂质分为5类,将定量构效关系(Quantitative Structure Activity Relationships, QSAR)评估作为遗传毒性杂质风险评估的第一步;
2006年,EMA发布首个正式指南《Guideline on the Limits of Genotoxic Impurities》,2008年由CHMP安全工作组(SWP)发布Q&A,提出决策树;
2008年,FDA发布《Genotoxic and Carcinogenic Impurities in Drug Substance and Products: Recommended Approach》;
2017年,ICH发布指南《M7(R1) Assessment and Control of DNA Reactive(Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic》;
2020年,中国药典2020版四部通则9306 遗传毒性杂质控制指导原则随药典发布正式公开。
随着相关科学研究的深入,已发布的指南也会逐步优化调整,继续协调企业与监管机构之间的不同意见。
三、风险评估过程
遗传毒性杂质的研究是综合学科,涉及有机化学(产生和清除机理)、生物化学(致突变致癌机理)[4]、毒理学(体内体外遗传毒性试验、致突变和致癌性数据评估等)、计算机分子模拟(软件预测)、制药工艺(合成路线筛选、遗传毒性杂质的工艺控制等)、分析化学(产品检测)等;因此,在ICH指南中不属于Q3系列,归类在M系列下(Multidisciplinary Guidelines)。原料药或制剂产品中遗传毒性杂质的风险评估过程也会涉及上述各学科知识。同样的,监管机构审评过程中涉及到非临床安全性数据(实验数据、计算机预测数据及相关文献可靠性评估、AI和PDE计算等)部分由药理毒理专业审评;确定AI或PDE后,结合每日最大给药剂量和用药周期确定的限度、相关的分析检测方法学和工艺控制策略由药学专业审评。
对遗传毒性杂质进行风险评估时,需考虑到产品所处的不同开发阶段。处于临床I期的产品和申报上市的产品,在遗传毒性杂质的工艺控制、限度确定、检测方法开发等要求上是不同的,大致可按如下风险评估流程进行:
第一步,列出产品工艺路线中的所有可能杂质,不限于起始物料、中间体、试剂、催化剂、溶剂、可能的反应副产物(尤其是最后一步反应)、降解产物(来自强制降解、长期和加速稳定性试验、制剂工艺过程或原辅料相容性试验)等;代谢产物和制剂降解产物需要另行考虑,这些杂质大部分时候也是中间体或起始物料,在原料药合成路线设计时就需要考虑到可能产生遗传毒性杂质的代谢途径和降解途径,并尽量避免。代谢产物和制剂降解产物的研究控制在《Genotoxic Impurities:Strategies for Identification and Control》[3]一书第6章节和第15章节有详细阐述,此处不再赘述。
第二步,采用数据库/文献检索、QSAR评估、体外遗传毒性试验(通常是细菌回复突变试验)、体内遗传毒性试验等方法对上述化合物进行遗传毒性结构评估;充分利用可靠文献和软件预测进行评估,尽量减少动物试验。按照标准流程进行的细菌回复突变试验需要至少300mg各化合物,某些难以获得的杂质用于该试验可能比较困难。基于结构的评估可以有效预测细菌回复突变试验结果,即上述的可靠的文献检索(致癌性和致突变性毒理学数据)和QSAR评估。
1)文献检索
可靠的化合物致癌性和致突变性毒理学文献检索首先来源于可靠的数据库或专业网站上经过各学科专家组织充分评估过的与普通人群暴露(食物、水、空气等)相关的安全性数据。遗传毒性杂质在药学领域是较新的概念,但是相关化合物在环保、化学危险品、人类健康等领域已有多年毒理学研究数据积累。以下是常用的数据库或检索网站:
IARC(International Agency for Research on Cancer,国际癌症研究署)是世界卫生组织下属的一个跨政府机构,进行世界范围内的癌症流行病学调查和研究工作,从1971年起组织专家组收集和评价世界各国有关化学物质对人类致癌危险性的资料,其分类网站(https://monographs.iarc.fr/list-of-classifications)定期更新化合物致癌性分类,最新的分类标准详见表1。该分类与遗传毒性杂质的分类不同,只针对致癌性进行分类,ICH M7中的遗传毒性杂质基于致癌性和致突变性进行分类。
表1 IARC化合物致癌性分类
| 类别 | 定义 | 分类标准 |
| 1类 | 对人具有致癌性 | 有足够的证据证明对人类具有致癌性;人类暴露有强有力的证据,同时在实验动物中显示出重要的致癌物特征和足够的致癌性证据。 |
| 2A类 | 对人很可能是癌 | 进行至少两次下列评价,包括至少一次涉及人体和人体细胞或组织的评价:1.人类致癌性证据有限;2.实验动物有足够的致癌证据;3.强有力的证据显示具有致癌物质的关键特征;这类物质或混合物对人体致癌的可能性较高,在动物实验中发现充分的致癌性证据,对人体虽有理论上的致癌性,但实验性的证据有限。 |
| 2B类 | 对人可能致癌 | 该类别存在下列评价之一的情况:1.人类致癌性证据有限;2.动物实验中有足够的致癌证据;3.强有力的证据表明具有致癌物关键特征(无论是暴露于人类还是人体细胞)。 |
| 3类 | 对人的致癌性尚无法分类 | 不属于以上任何类别的因素通常被放在这个类别中。当在动物实验和人类致癌性证据均不足时,通常放在此类别;当有强有力的证据表明在实验动物中有致癌性机制但不能在人类身上起作用,在人类身上的证据还不够时,也可放在此类别中。 |
ToxInfo(毒理学数据网络,https://www.toxinfo.io/)是美国国立医学图书馆(National Library of Medicine, NLM)建立的门户网站,由涉及毒理学、危险化学品、环境卫生等相关领域的数据库构成,由NLM的专业信息服务部执行的毒理学和环境卫生信息计划来管理。ToxInfo提供了对下列数据库的免费访问检索:HSDB、ChemIDplus、CTD、CPID、Haz-Map、IRIS、ITER、CCRIS、CPDB、GENE-TOX。其中我们常用的是CPDB、IRIS、CCRIS、GENE-TOX、HSDB、ITER。
CPDB(Carcinogenic Potency Database,致癌性数据库),由加利福尼亚大学和劳伦斯伯克利实验室开发,收录了自1950年代以来进行的6540项慢性、长期动物癌症实验结果的标准化分析,只提供1980~2011年间的信息,现已不再更新。CPDB收录的毒理学数据(鼠伤寒沙门氏菌Ames试验结果、不同种属动物的TD50等)被各国药监机构广泛认可,可直接使用,一般选择最低的TD50值进行AI(Acceptable Intakes,可接受摄入量)值计算。
IRIS(Integrated Risk Information System, 综合风险信息系统)是美国环境保护署(Environmental Protection Agency, EPA)开发的一个人类健康评估计划,评估暴露于环境污染导致对于健康的影响(癌症和非癌症)的情况,收录的化学品信息都经过了EPA科学家们的审定,并代表了EPA的结论性意见。IRIS除了对特定化合物的不同致癌性和致突变性毒理学试验结果进行专业评估汇总,也会提供明确的用于计算参考剂量(Reference Dose, RfD)的NOEL(No-Observed Effect Level,未观察到作用水平)、NOAEL(No-Observed Adverse Effect Level,未观察到有害作用水平)或LOEL(Lowest-Observed Effect Level,观察到作用的最低水平)、相应于1/100000风险水平的饮用水限度或吸入剂量等数值,可用于计算可接受摄入量,权威性和认可度较高。
CCRIS(Chemical Carcinogenesis Research Information System,化学致癌研究信息系统)由美国国立癌症研究所(National Cancer Institute,NCI)开发和维护,收录了9000多种致癌性、致突变性、诱发肿瘤和抑制肿瘤的试验结果的化学品记录,只提供1985~2011年间的信息,现已不再更新。CCRIS罗列文献发布的致癌性和致突变性简单试验过程和试验结果(阴性或阳性),未提供直观的TD50、NO(A)EL、LOEL等数据,这些试验结果经常不一致,需要专业的毒理学人员对试验过程合理性进行判断,质量研究人员只能参考。
GENE-TOX(Genetic Toxicology Data Bank, 遗传毒理学数据库)由美国环境保护署创立,收录3200多种化学品的经专家审评的遗传毒理学试验(含致突变性)结果,只提供1991~1998年间信息,现已不再更新。同CCRIS一样,只罗列简单的动物试验结果,仅供参考。
HSDB(Hazardous Substances Data Bank, 危险物质数据库)收录了5800多种危险化学品的毒理学数据,提供了充分的参考文献,并且经过科学审查小组的评审。Section 5下提供的毒理学试验总结中也能找到NO(A)EL、LOEL值,可靠性上可能不如IRIS提供的结果,仅供参考。
ITER(International Toxicity Estimates for Risk,国际毒性风险评估)提供来自全球各权威机构的600多种环境关注化学品的健康风险值和癌症分类,用表格形式对比不同机构(CDC/ATSDR、Health Canada、RIVM、 US EPA、IARC、NSF International等)的数据差异,并作出解释,提供来源文献和更详细的说明链接。我们可以在这个数据库中找到化合物的各类权威毒理学试验结果,并直观看到数据比较,最终选择哪个数据进行限度计算还需要专业毒理学人员充分合理评估。
ToxInfo提供的上述数据库,没有毒理学专业知识的质量研究人员用的比较多的还是CPDB和IRIS,能获得直观的TD50计算AI或NO(A)EL等计算PDE。
其他可靠网站和数据库还有:Carcinogenicity Database (Lhasa Ltd.)、ATSDR、IPCS、CalEPA、NTP等。
Carcinogenicity Database(https://carcdb.lhasalimited.org/carcdb-frontend/)由Lhasa Ltd.公司开发,提供比CPDB更加详细的TD50值获得的试验过程。Lhasa Ltd.公司也开发了Derek、Meteor、Vitic和Zeneth等预测软件,其中Derek被药监机构广泛认可。
ATSDR(Agency for Toxic Substances & Disease Registry,有毒物质和疾病登记局)是美国健康和公共服务部门下属的联邦公共健康机构,预防与有毒物质有关的有害暴露和疾病,提供经专家小组评估过的毒理学数据汇编(Toxicological Profiles),该网站下(https://www.atsdr.cdc.gov/mrls/mrllist.asp#169tag)定期更新有毒物质的不同给药途径下的急慢性动物试验结果MRLs(Minimal Risk Levels,最小风险剂量)值,类似于EPA提出的参考剂量(RfD),是无健康风险的暴露剂量的估算值,具体是指人类在确定的时间内每日接触有害物质而不产生明显的非致癌性损害作用的剂量。该值可靠性高,可计算PDE值。
IPCS(International Programme on Chemical Safety,国际化学品安全规划小组)由世界卫生组织、国际劳工组织和联合国环境规划署共同成立的有关化学品安全的国际合作机构,开展化学品对人体健康和环境风险的评价,包含动物试验结果、人体和环境暴露及安全性评价,其网站也会公布特定化合物的CICAD(Concise International Chemical Assessment Document, 简明国际化学品评估文件)及可接受摄入量(https://www.who.int/ipcs/publications/cicad/cicads_alphabetical/en/),国际认可度高,可参考使用。
CalEPA(California Environmental Protection Agency, 加利福尼亚州环境保护局)提供毒理学试验结果和直观的口服或吸入斜率因子(Slope Factor)和NSRL(No Significant Risk Level,无显著风险水平),可直接作为特定化合物的可接受摄入量。网址链接:https://calepa.ca.gov/,搜索栏中直接输入CAS号即可。
NTP(National Toxicology Program,美国国家毒理计划),提供各化合物的毒理学研究结果,网址链接:https://ntp.niehs.nih.gov/,搜索栏中直接输入CAS号即可。
上述各数据库或网址均是被国际广泛认可的权威性安全性数据检索来源,某些化合物相关研究较多,各网站均能提供相关毒理数据,此时需要对不同网站检索到的毒理学数据进行试验过程合理性评估,最好由毒理学专业人员进行。某些化合物所能检索到的毒理学数据较少,当上述网站都没有相关信息时,可尝试在google中检索“CAS号 EPA”,也能获得US EPA公布的信息。
文献检索和结果分析需要一定时间,大量的英文文献和毒理学专业术语可能有些费劲,对安全性数据的评估也需要毒理学专业人员参与,此时采用QSAR方法对化合物进行结构评估就显得简单一些。
2)QSAR评估
ICH M7推荐采用2种互补机理的QSAR毒性预测方法,如基于专家规则的Derek(Lhasa Ltd.开发, FDA、EMA推荐使用)、Compact、OncoLogic等;基于统计学的Sarah、Topkat (Accelrys Inc.)等。上述软件的毒理学数据均是来自化合物实际Ames试验结果,通过专家系统知识或已有的毒理学数据统计分析,建立模型对待测物进行毒性预测。软件预测的灵敏度(sensitivity)和特异性(specificity)在苯胺或氮杂芳胺类遗传毒性杂质的评估上有一定缺陷,不过会随着实际化合物Ames试验结果和更精准的警示结构的不断补充逐步提高,使用者需特别关注这类杂质的评估结果。企业也可以采用自行开发的软件进行毒性评估,但所采用的方法需遵循经济合作与发展组织(OECD)制定的一般验证原则[5],且应使用2014年M7发布之后开发的模型进行预测。
如果经2种互补的QSAR方法预测均没有警示结构,可以确定该化合物没有致突变性,可以归类为遗传毒性杂质分类列表中的第5类杂质。如果长期给药的杂质日摄入量大于1mg,不管分类如何,都应进行最低筛选要求的遗传毒性试验(点突变和染色体畸变)。任何阳性或相互矛盾的结果可以通过专业知识(与具有细菌回复突变试验数据且结构相似的类似物比较、由专家对化学结构进行评价以确定化合物是否具有DNA反应性、采用相同方法的其他经验证的QSAR模型论证等)或开展遗传毒性试验进行充分合理论证。当软件评估的结果与药监机构或ICH等权威国际组织发布的数据或Ames试验结果冲突时,以药监机构和权威国际组织公布的结果或Ames试验结果为准。
3)遗传毒性试验评估
遗传毒性试验方法有多种,根据试验检测的遗传终点,可将检测方法分为3大类,即基因突变、染色体畸变、DNA损伤;根据试验系统,可分为体内试验和体外试验。体外试验通常有细菌回复突变试验(即Ames试验,推荐使用,至少应包含5种菌株组合:TA98、TA100、TA1535、TA1537或TA97或TA97a、TA102或大肠埃希杆菌WP2 uvrA或大肠埃希杆菌WP2 uvrA(pKM101),没有特殊说明时,一般为鼠伤寒沙门氏菌)、体外中期相染色体畸变试验、体外微核试验、小鼠淋巴瘤L5178Y细胞Tk基因突变试验(MLA)等;体内试验通常有转基因突变试验、Pig-a试验(外周血)、微核试验(外周血和骨髓)、大鼠肝脏非程序性DNA合成(UDS)试验、碱性彗星试验等。上述各试验的标准方法详见ICH S2 《人用药物遗传毒性试验和结果分析指导原则》[6]。
QSAR结果不明确或者为3类时,可以进一步开展细菌回复突变试验或其他体外遗传毒性试验;体外结果为阳性时,可以进一步开展体内遗传毒性试验,明确体内致突变风险,指导限度制定。其他体内外遗传毒性试验的选择应根据杂质的反应机理和预期靶组织暴露的知识进行科学论证,且试验设计应参考ICH S2 《人用药物遗传毒性试验和结果分析指导原则》[6]。
第三步,当完成上述文献检索或软件评估过程后,需要根据检索/评估的结果,即是否具有明确的致突变和致癌性数据,对各化合物进行分类,具体分类标准和M7推荐的控制策略见表2[2]。
表2 ICH M7 化合物致癌性和致突变性分类
| 分类 | 定义 | 拟定的控制措施 |
| 1 | 已知致突变致癌物 | 控制不超过该化合物特定的可接受限度 |
| 2 | 致癌性未知的已知致突变物(细菌致突变阳性,但无啮齿动物致癌性数据) | 控制不超过可接受限度(合适的TTC) |
| 3 | 有与原料药结构无关的警示结构,无致突变数据 | 控制步超过可接受限度(合适的TTC)或进行细菌只突变试验;无致突变性,归为5类;有致突变性,归为2类 |
| 4 | 有警示结构,且与经测试无致突变性的原料药及其相关化合物(例如,工艺中间体)具有相同的警示结构 | 按非致突变杂质控制 |
| 5 | 无警示结构,或虽有警示结构但有充分的数据证明无致突变性或无致癌性 | 按非致突变杂质控制 |
上述不同类别遗传毒性杂质可接受摄入量计算方式不同,一般可用法规公布的数据、以线性机制为基础的AI计算方法、以阈值机制为基础的PDE(Permitted Daily Exposure,每日允许暴露量)计算方法、TTC和分期TTC(属于AI计算方法的一种)、其他安全性数据等计算获得。
1)法规公布的限度
ICH M7公布了部分化合物的可接受摄入量[2],详见表3。FDA公布了亚硝胺类化合物的可接受摄入量[7],详见表4。
表3 ICH M7中的部分化合物可接受摄入量(2-1)
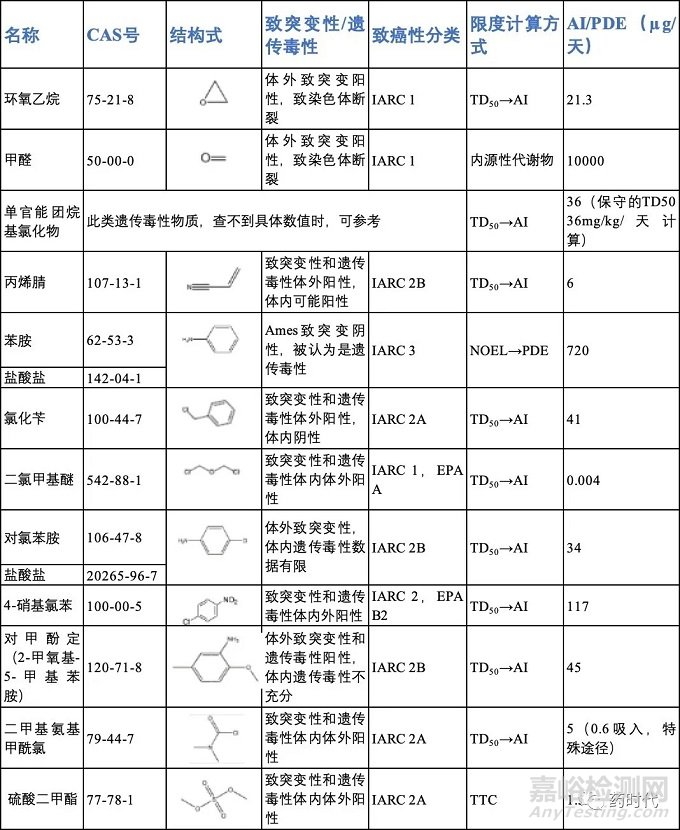
表3 ICH M7中的部分化合物可接受摄入量(2-2)
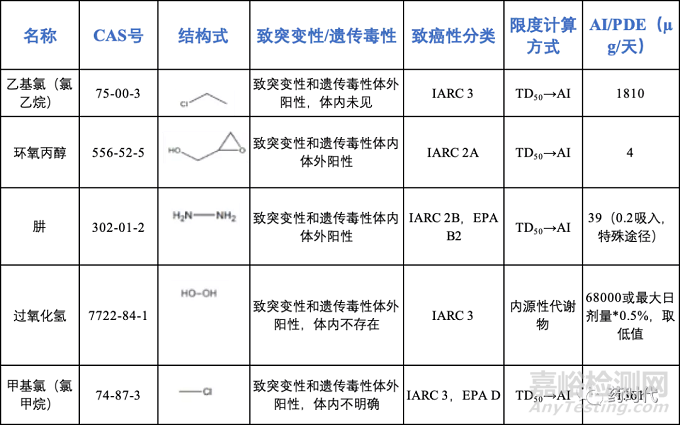
表4 FDA公布的亚硝胺类化合物可接受摄入量
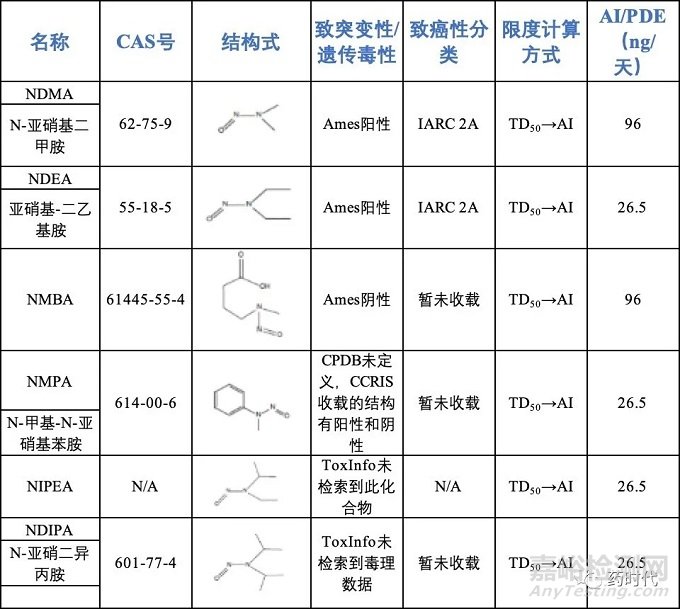
注:当只含有1个N-亚硝胺类杂质时采用上述AI值计算限度,
含有2个及以上时总AI值应为26.5ng/天。
2)AI计算方法
如果杂质具有足够的致癌性数据,且非阈值机制,可以采用啮齿类动物的TD50值通过线性外推计算化合物的AI值。TD50可从CPDB数据库或第二步文献检索中直接获得,也可使用与CPDB相同方法从已发表的研究中自行计算获得,ICH M7文末举例了具体的计算方式[8-9]。TD50的1/50,000相当于终生潜在发生肿瘤的风险为十万分之一。
3)PDE计算方法
PDE的计算在残留溶剂和元素杂质中就有广泛运用,当杂质致突变致癌作用机制为阈值机制时,可通过NO(A)EL、LOEL和不确定因子来计算PDE值。
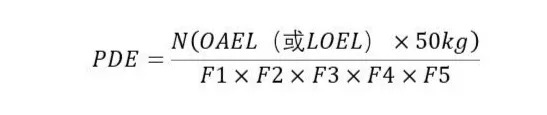
式中,F1为从不同物种外推到人的因子;
F2为个体差异因子;
F3为根据毒性暴露周期采用的可变因子;
F4为根据毒性严重情况采用的可变因子;
F5采用NO(A)EL时,一般为1,采用LOEL时,根据毒性严重程度确定,最高为10。
PDE的具体计算在ICH Q3C和Q3D中有详细举例,式中参与计算的NO(A)EL和LOEL值单位为mg/kg/天,采用第二步文献检索时,有时候获得的NO(A)EL和LOEL值为吸入研究的结果,单位是ppm,需要结合种属体重和呼吸量转换为mg/kg/天。F1~F5的具体数值需要结合获得NO(A)EL和LOEL值的长期(慢性)动物试验过程来确定。此处注意大鼠(rat)和小鼠(mouse和mice)的区别。进行第二步文献检索时,可在IRIS、HSDB、ITER等检索到NO(A)EL和LOEL值。
有时候NO(A)EL和LOEL值较难获得时,也有采用LD50(50%动物致死量)计算NOEL值的情况存在,具体公式如下:
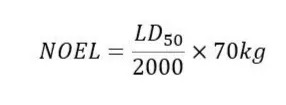
这个公式来自于APIC《Guidance on Aspects of Cleaning Validation in Active Pharmaceutical Ingredient Plants》[10],式中NOEL单位为mg/天,70kg为欧洲人体重,2000为口服剂型的原料药安全系数,获得的NOEL值进一步用于MACO(Maximum Allowance Carryover,允许最大残留)计算,而不是PDE计算。LD50为急性毒性试验结果,NOEL为长期(慢性)毒性试验结果,D.W.Layton等研究过从LD50→NOEL的可行性[11],但是LD50→NOEL→PDE的过程在残留溶剂、元素杂质、遗传毒性杂质等的可接受摄入量的计算中本身存疑较多,不建议使用。
目前一般认为,致突变杂质直接引起DNA损伤,以线性机制为主,因此可接受摄入量计算更多还是以AI、TTC等为主,对阈值机制的研究较少,PDE计算方式的使用需要更多毒理学评估,明确阈值机制存在。
4)其他安全性数据计算可接受摄入量
除了TD50、NO(A)EL、LOEL等毒理学数据,在第二步文献检索时也能获得化合物相应于十万分之一风险水平的饮用水限度(IRIS)、吸入剂量(IRIS)、MRL(ATSDR)、NSRL(CalEPA)、BMDL10等可直接或进一步计算获得最终的可接受摄入量。注意,应以最新且具有科学支持的数据作为计算基础。
上述各数据推导可接受摄入量过程举例如下:
①当IRIS公布的某化合物相当于1/100,000风险水平的饮用水限度为0.1μg/L时,日饮用水量为2L/天(设定值),可接受摄入量为0.1μg/L×2L/天=0.2μg/天;
②当IRIS公布的某化合物相当于1/100,000风险水平的吸入暴露量为0.8μg/m3时,日呼吸量为20m3/天(设定值,有时候采用28,800L/天),可接受摄入量为
0.8μg/m3×20m3/天=16μg/天;
③采用MRL计算PDE:
PDE=MRL(mg/kg/天)×50kg,
或PDE=MRL(mg/m3)/1,000m3/L×28,800L/天
校正因子已经包含在MRL值中,PDE计算不需要安全系数。
用于元素杂质的可接受摄入量计算需要区分不同给药途径的MRL值,但是遗传毒性杂质除非存在特定给药方式问题(如肿瘤的部位特异性或化合物的强效接触位点致癌性),否则无需根据给药途径调整可接受摄入量,一般首选长期/慢性毒性试验得到的最低的MRL值。
④NSRL(如40μg/天)可以直接作为可接受摄入量;
⑤查阅的BMDL10可以直接除以10,000线性外推至十万分之一风险发生率。
5)TTC或分期TTC
对于无毒理学研究数据的杂质可以采用TTC(单个杂质,1.5μg/天)作为可接受摄入量。TTC是从最敏感物种和肿瘤发生最敏感部位获得的TD50(1.25mg/kg/天)线性外推至十万分之一风险发生率的剂量,通用于大部分药物。如果有2个2类或3类杂质,应制定每个杂质的可接受摄入量;如果质量标准中有3个及以上的2类或3类杂质,应按表5控制总可接受摄入量。其中1类杂质和制剂中形成的降解产物应单独控制,不计入总可接受摄入量。
短于终生给药的药品中致突变杂质的TTC值可调整为更高剂量,即1.5μg/天累积70年的总量(38.3mg)平均分配到短期的总给药天数中,具体调整见表5。间歇给药时应根据总给药天数计算。
表5 单个杂质和总杂质的TTC和分期TTC
| 治疗期 | ≤1个月 | >1~12个月 | >1~10年 | >10年~终生 |
| 单个杂质TTC(μg/天) | 120 | 20 | 10 | 1.5 |
| 多个杂质总TTC(μg/天) | 120 | 60 | 30 | 5 |
从TD50值推导的AI值,也可以根据给药周期,按照表5的比例同等放大短期给药的可接受摄入量,但不超过0.5%。举例,如果终生暴露时,某化合物可接受摄入量为15μg/天,短于终生暴露时限度可增加至100μg/天(>1~10年治疗时长),200μg/天(>1~12个月治疗时长)或1200μg/天(<1个月治疗时长)。但是,如果最大日服用剂量为100mg,则<1个月时长的每日可接受摄入量应为0.5%×100mg=500μg,而不是1200μg。此方法不适用于阈值机制的PDE放宽数值,但是对于短期暴露(≤30天),具体问题具体分析后,可以提高杂质可接受摄入量。
6)其他情况
对于与已知的某类致癌化合物在化学结构上相似的杂质,充分论证相似理由(结构相似,致癌致突变机理相似)后,可以按相似化合物的可接受摄入量计算。
如果某杂质通过其他途径(食品、内源性代谢物等)在人体中的暴露量更大,可接受摄入量可以提高;致突变代谢产物的限度制定通常需要结合体内遗传毒性研究结果制定,致突变制剂降解产物限度制定通常取决于长期稳定性试验结果。
参考药典收载的其他品种某遗传毒性杂质化合物的限度时需要注意,不可直接使用,应考虑该品种的给药周期和日服用剂量是否与待计算品种一致。
从上述各途径获得的杂质可接受摄入量(AI、PDE、TTC等)计算限度如下:

式中,药品最大日服用剂量来自药品说明书。
第四步,按上述步骤筛选出来的遗传毒性杂质或潜在遗传毒性杂质,应分析其在API、中间体或制剂产品中残留的可能性。遗传毒性杂质的亲电性使得它们一般具有一定化学反应活性,详见表6[3]。在API合成工艺中,这些杂质可能在后续反应步骤中去除,也可能反应生成其他物质,最后残留在产品中的可能性较低。不过药监机构不太认可纯理论分析,在杂质控制策略中可以结合清除因子和检测结果证明产品或中间产品中的杂质残留情况。
表6 遗传毒性杂质按化学反应活性分类
| 化学反应活性 | 遗传毒性杂质结构类别 |
| 高活性 | 环氧化物、醛类、磺酸酯类、肼类、酰基卤化物、氮杂环丙烷 |
| 中等活性 | 硫芥类、氮芥类、迈克尔反应受体、卤代烯烃、 |
| 低活性 | 嘌呤、嘧啶、氨基甲酸酯类、芳香胺、硝基化合物 |
注:高活性指极易与亲核试剂反应。
为了量化分析杂质残留可能性,将杂质的物理化学性质和工艺过程结合起来,提出了清除因子(表7)的概念,数值越大,代表工艺清除的可能性越高。特定杂质在特定工艺流程中的总清除因子为各步清除因子的乘积。当清除因子大于10000时,杂质在API中的残留可能性极低,除非每日最大剂量在1g以上,否则可以不做相关控制;当清除因子在100~10000之间时,需要做加标实验证明工艺路线的清除能力;当清除因子在100以下时,说明杂质在最后或者倒数第二步引入,需要谨慎对待,并严格控制。[3,12]
表7 物理化学性质与清除因子
| 物理化学性质 | 清除因子 |
| 化学反应活性 | 高活性=100中等活性=10低活性=1 |
| 溶解性 | 易溶=10溶解=3略溶或不溶=1 |
| 挥发性 | 杂质沸点比溶剂沸点低20℃以上=10杂质沸点在溶剂沸点的±10℃范围内=3杂质沸点在溶剂沸点的20℃以上或者不挥发=1 |
| 电离度 | 杂质的潜在电离度与API或基质明显不同=10 |
| 柱层析 | 杂质先于API洗脱=100杂质后于API洗脱=10 |
| 重结晶 | 杂质在重结晶溶剂中易溶=100杂质在重结晶溶剂中略溶=1 |
| 其他工艺流程,如树脂处理 | 具体情况具体分析 |
第五步,可能存在于API/制剂中的遗传毒性杂质,需要开展遗传毒性试验确认,通常是Ames试验,或者直接在各步控制在特定限度以下。
Ames试验结果为阴性时,按照普通杂质控制;Ames结果为阳性,但是杂质在产品或中间产品中能控制在特定限度以下,也可以接受;Ames结果为阳性,杂质在限度以上,可以进行体内遗传毒性试验,体内结果为阴性,按一般杂质控制;也可以重新评估工艺和路线,将杂质控制在限度以下;如果工艺控制不能将杂质水平降低至可接受限度以内,而杂质是ALARP水平,可以根据风险/收益分析合理提高杂质限度。
基于对产品和工艺的理解,以及风险管理原则,可将杂质控制点向上游移动,尽量减少终产品检测。ICH M7推荐4种API工艺杂质控制方法[2]:
方法1:定入API质量标准(常规检测或定期检测);
方法2:工艺上游控制-定入起始物料或中间体的内控标准;
方法3:工艺上游控制-定入起始物料或中间体的内控标准或过程控制,限度适当放宽,但是需要明确杂质去向和清除信息,并确认API中杂质始终控制在30%可接受限度内;
方法4:无需制定标准-明确工艺参数及其对残留杂质水平的影响,确信API中杂质存在风险可忽略不计(如1%TTC[5])。方法4适用于自身不稳定或工艺早期引入的可被有效清除的杂质,其可接受性由药监机构根据具体问题具体分析。
对于制剂降解杂质的控制,ICH M7建议根据降解试验(加速试验、光稳定性试验、强制降解试验、原辅料相容性试验等)评估降解途径与原料药和制剂的生产工艺和储藏条件的相关性,采取相应措施尽量控制致突变降解杂质产生和增长,杂质限度制定取决于长期稳定性试验结果。
各企业对遗传毒性杂质的研究和控制,可分类进行。同一类别杂质的致突变致癌作用机制、产生和清除机理、可接受摄入量制定、分析检测方法等有一定相似性,在项目运行过程中收集各品种的各类遗传毒性杂质信息,逐步建立企业自己的遗传毒性杂质库,在后续同类别遗传毒性杂质研究和控制中可以节约时间成本。
四、结语
在遗传毒性杂质的实际风险评估过程中,目前还大量存在根据文献[13]公布的警示结构“目测”化合物(大部分时候所列出的杂质谱清单不齐全)是否具备潜在的遗传毒性,没有进行可靠文献检索和QSAR评估,根据“目测”的结果,结合用药周期和最大日服用剂量,采用TTC或分期TTC计算出杂质限度,开发相关方法,检测多批次样品,根据结果作出相应的工艺调整,如果调整不了,寻求放宽限度的方法以使产品合格,完成遗传毒性杂质评估。这种评估方式在欧美申报中是不被认可的,在国内申报中也将逐步退出历史舞台。
参考文献
1. 中国药典2020版四部通则9306 遗传毒性杂质控制指导原则.
2. ICH Harmonised Guideline: Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk(R1).2017.
3. Andrew Teasdale, et al. Genotoxic impurities strategies for identification and control. John Wiley & Sons, Inc.,2010.
4. Romualdo Benigni, et al.Mechanisms of chemical carcinogenicity and mutagenicity:A review with implications for predictive toxicology. Chemical Reviews 2011;111:2507-2536.
5. ICH Harmonised Guideline: Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk(R1), Questions and Answers. 2020.Version 29.
6. ICH Harmonised Guideline: Guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use(R1).2011.
7. FDA Guidance for industry: Control of nitrosamine impurities in human drugs.2020.
8. David W. Gaylor, et al. Quick estimate of the regulatory virtually safe dose based on the maximum tolerated dose for rodent bioassays. Regulatory Toxicology and Pharmacology 1995; 22:57-63.
9. Charles Sawyer, et al. Calculation of carcinogenic potency from long term animal carcinogenesis experiments. Biometrics 1984; 40:27-40.
10. Active Pharmaceutical Ingredients Committee (APIC). Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants. 2016.
11. D.W.Layton, et al. Deriving allowable daily intakes for systemic toxicants lacking chronic toxicity data. Regulatory toxicology and pharmacology 1987; 7:96-112.
12. Nevenka Kragelj Lapanja, et al. Theoretical purge factor determination as a control strategy for potential mutagenic impurities in the synthesis of drug substances. Acta Chimica Slovenica 2017; 64:1-14.
13. Romualdo Benigni, et al. Development of structural alerts for the in vivo micronucleus assay in rodents. JRC Scientific and Technical Reports 2009.

来源:上海博志研新


