您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-03-26 15:45
导读
小分子共价抑制剂(covalent inhibitors)也称不可逆抑制剂(irreversible inhibitors):是通过共价键与靶蛋白残基发生不可逆结合,从而发挥其生物学功能的一类抑制剂。一般来说,共价抑制是一个两步过程(图1a)。首先,抑制剂可逆地与靶酶结合,从而将小分子中的富含电子的化学弹头近距离接近酶中的活性残基。在第二步,抑制剂的两个反应实体和酶之间发生反应形成共价键。常见的共价靶头有丙烯酰胺、环氧、氯乙酰基以及氧磺酰氟和磺酰氟等等(图1b),常见的氨基酸活性残基有半胱氨酸、丝氨酸、酪氨酸、赖氨酸以及精氨酸和谷氨酸等等(参考文献1)。
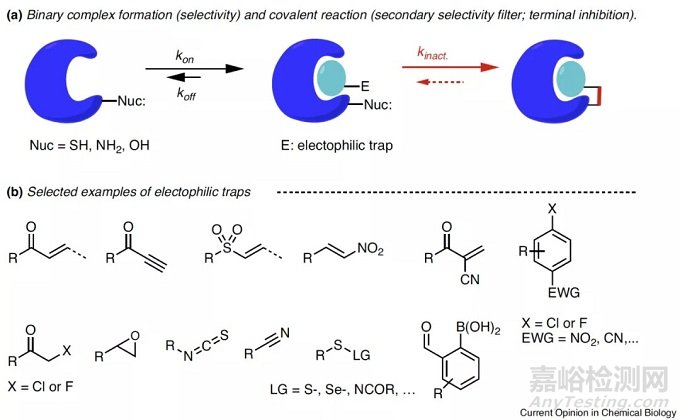
图1 共价抑制剂发挥功能的过程以及常见共价靶头
相对可逆抑制剂来说,共价抑制剂具有较高的生化效率、作用较强并且持久,给药剂量以及给药频率减少、药效学与药代动力学分离,药物被迅速清除也能够维持效力、能够预防耐药性的产生、能够靶向特定蛋白罕见的、非保守残基,达到较高的选择性等特点,但是共价抑制剂存在脱靶导致的副作用。本文将重点介绍蛋白激酶、RAS蛋白以及其它被广泛研究的靶蛋白相关的共价抑制剂的研究进展,进而阐述设计共价抑制剂的挑战和建议。
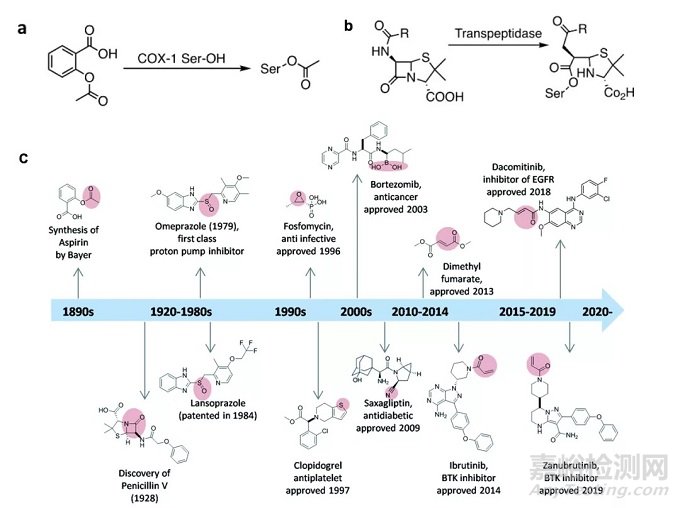
图2 共价药物发现的简要时间线
最初发现的共价抑制剂是乙酰水杨酸和青霉素。乙酰水杨酸的乙酰基团可以共价连接到COX-1上的丝氨酸进而抑制活性;青霉素中的β-内酰胺可以和转肽酶中活性位点丝氨酸发生共价结合。近年来共价抑制剂的发展史如图2所示。(参考文献2)
蛋白激酶类的共价抑制剂
激酶可以将ATP上的γ-磷酸基团转移到特定靶分子中的氨基酸残基上,从而改变蛋白质、酶的构象和活性。到目前为止,已经被发现了518种蛋白激酶,这些蛋白激酶的三维结构具有一定的相似性,都有ATP结合domain,因此可以对非共价抑制剂进行合理的结构优化后引入共价靶头从而设计高效的共价抑制剂。
表皮生长因子受体(EGFR)的不可逆共价抑制剂
EGFR属于受体酪氨酸激酶家族,通过催化蛋白酪氨酸磷酸化控制信号转导。表皮生长因子受体(EGFR)是一种细胞表面蛋白,可以结合其天然的配体进而诱导酪氨酸自磷酸化和信号细胞增殖。该基因的特定突变与非小细胞肺癌有关(NSCLC)。最常见的是19外显子缺失(DE746-A750)和一个单点突变,L858R。这些被称为“激活突变”,因为它们导致配体非依赖的酪氨酸激酶活性。
非正常升高的激酶活性可以被ATP竞争性可逆药物抑制,如吉非替尼和厄洛替尼可以抑制激酶活性水平(图3a)。然而,另一个活性位点突变T790M降低了它们的至少50%的功效。近年来报道了一些不可逆抑制剂如PD168393,PF00299804 (dacomitinib)、和EKB569 (pelitinib)(图3b)均有报道,但并不能成功克服与T790M突变相关的药物疗效问题。Neratinib可以和C797进行共价结合,但由于其生物利用度不高以及腹泻的副作用而没有进一步的研究。Afatinib是一种ErbB家族抑制剂剂,是FDA批准的第一个EGFR共价抑制剂。单独使用阿法替尼延长无进展生存(PFS)时间几乎是安慰剂联合治疗患者的3倍,当使用西妥昔单抗联合治疗,对L858R / T790M erlotinib-resistant肿瘤也产生不错的效果,但会引起皮疹和腹泻的副作用。

图3 EGFR可逆和不可逆抑制剂以及部分不可逆抑制剂和蛋白的共晶结构
为了减轻副作用,科学家们设计了三代共价抑制剂,WZ4002可以与C797形成共价连接,且嘧啶环上的氯可以与790位甲硫氨酸形成卤键,这个相互作用增加了化合物对EGFR(T790M)的选择性。此外2015年11月,FDA批准了由阿斯利康开发的TAGRISSOTM(奥希替尼,原名AZD9291)图4b),用来治疗晚期NSCLC。临床前研究表明IC50值为对L858R突变体的活性为12 nM,对L858R/T790M EGFR双突变体的活性为1 nM,该药物对EGFR L858R/T790M表现出约200倍的效力。

图4 第三代EGFR不可逆共价抑制剂
Bruton’s tyrosine kinase (BTK)的不可逆共价抑制剂
布鲁顿酪氨酸激酶(Bruton’s tyrosine kinase,BTK)是胞浆内非受体型酪氨酸激酶TEC家族中的一员,在B细胞生长发育、增殖分化过程中起着重要作用,同时也是B细胞恶性肿瘤的治疗靶点,BTK突变导致一种免疫缺陷疾病—X-连锁无丙种球蛋白血症(X-linked agammaglobulinemia,XLA)。Ibrutinib(原名PCI32765,见图5a)是一种共价BTK抑制剂,它被批准用于治疗慢性淋巴细胞白血病(CLL)、套细胞淋巴瘤(MCL)和瓦尔登斯特伦氏巨球蛋白血症。Ibrutinib通过减少Y223残基上的自磷酸化来抑制BTK。其丙烯酰胺部分与C481反应形成BTK中的共价键激酶结构域,它将BTK锁定在变构抑制状态,虽然它在治疗B细胞淋巴瘤和白血病上很成功,ibrutinib会导致副作用,如出血,皮疹,腹泻,心房颤动。为了克服副作用,科学家们设计了几个第二代BTK抑制剂(图5d),其中一种抑制剂,阿卡拉替尼(acalabrutinib,图5c),显示效力强,口服吸收快,短半衰期以及减少与EGFR和其他Tec家族蛋白的结合。

图5 BTK共价抑制剂
不可逆的p90核糖体S6激酶不可逆共价抑制剂(RSKs)
p90核糖体S6激酶(RSK)家族是由丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路激活的一种蛋白激酶,在黑色素瘤生长和增殖中起关键作用。根据RSK非共价抑制剂PP1以及蛋白共晶结构,利用计算机设计,设计合成了RSK不可逆共价抑制剂FMK,意外发现FMK对大量激酶都有作用,此外2018年Gothelf, Nissen等人设计了化合物富马酸二甲酯,发现其是MSKs的变构共价抑制剂(图6)。
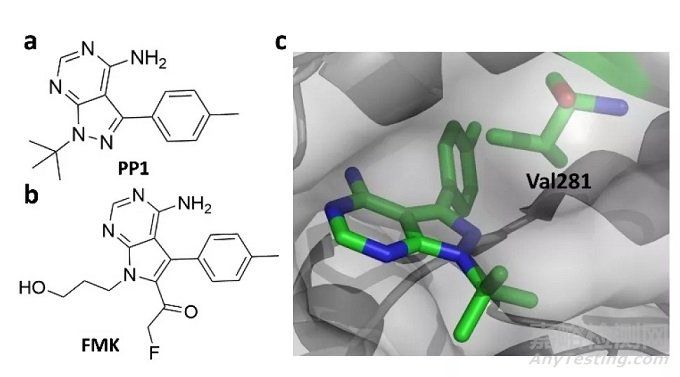
图6 RSK抑制剂
蛋白激酶的可逆共价抑制剂
可逆共价抑制剂的结构与作用机制与不可逆共价抑制剂均相似, 而不同的是其与目标靶蛋白的共价结合是可逆的(图7).可逆共价抑制剂的亲电弹头多为氰基、酮羰基等可逆亲核加成反应受体.而其与靶标共价结合的可逆性, 使得可逆共价抑制剂的药代动力学特征介于不可逆共价抑制剂和非共价抑制剂之间。可逆共价抑制剂在一定程度上继承了不可逆共价抑制剂作用时间长、有效浓度低等优点, 同时也减少了脱靶带来的毒性风险。

图7 可逆共价抑制剂的原理
Taunton及其同事成功地开发了高反应性的2-氰基丙烯酸酯基RSK2可逆共价抑制剂 (图8a),此外也有一系列可与BTK 的Cys481发生可逆结合的共价抑制剂(图8b)

图8 常见的可逆共价抑制剂
RAS家族的共价抑制剂
RAS癌基因参与人类肿瘤的发生发展,自82年Weinberg等人发现其中有活化的H-ras基因后,引起了人们对RAS癌基因在人类肿瘤发生、发展中所起作用的极大关注。经十余年研究认为,RAS癌基因参与细胞生长和分化的调控,参与多种肿瘤的形成与发展。迄今为止,对于RAS癌基因突变,还没有有效的治疗方法。然而,最近关于开发RAS蛋白的强效小共价抑制剂的报道又重新燃起了人们对这些所谓的“不可成药”蛋白质的希望
KRAS突变体的共价抑制剂
KRAS蛋白是由 KRAS 基因编码的一种小 GTP 酶,它属于 RAS 超蛋白家族,是细胞生长的重要调节蛋白,一旦 KRAS 被激活后,可以激活多条信号通路,促进细胞的增殖。
KRAS 是实体瘤中最常见的癌基因之一,大约 30% 的肿瘤都存在 KRAS 突变,包括 90% 的胰腺癌,50% 的结肠癌和 25% 的肺癌。最常发生基因突变的位点是第 12、13 和 61 位密码子,其中以 12 位密码子的突变最为常见。最近,Shokat和同事报道了一组半胱氨酸反应抑制剂,它们与KRAS G12C突变体相互作用,随后与G12C形成共价加合物(图9a),此外他们筛选了其它亲电试剂,例如丙烯酰胺和乙烯基磺胺,它们与G12C残基形成不可逆的共价键(图9c,e)。
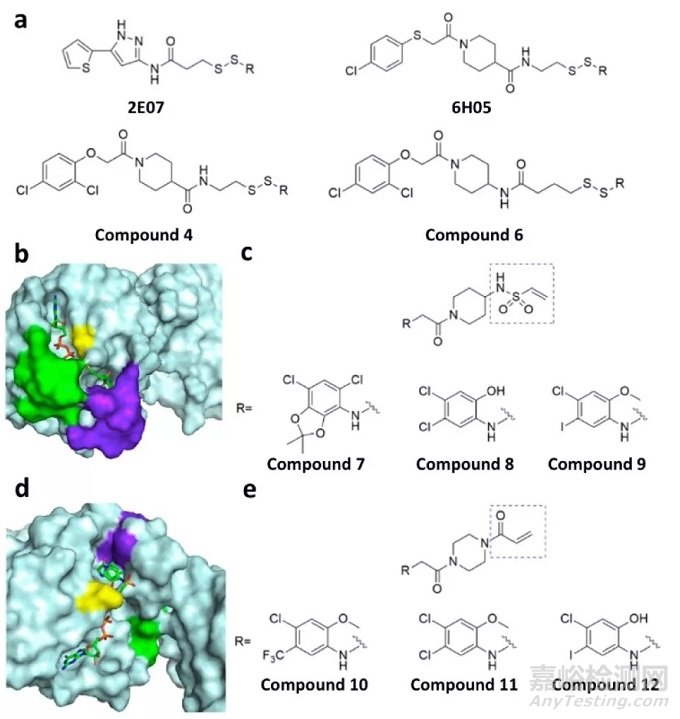
图9 KRAS突变体的共价抑制剂
其它蛋白的共价抑制剂
乙酰胆碱酯酶的共价抑制剂
乙酰胆碱是一种刺激胆碱能的神经递质,阿尔兹海默患者中乙酰胆碱受体降低,抑制乙酰胆碱酯酶(AChE)是目前治疗阿尔茨海默氏病(AD)最有效的方法。Rivastigmine可以共价的结合在乙酰胆碱酯酶上,从共晶结构上可以看出,S203(酯位)和E202(阴离子位)与化合物有相互作用,Rivastigmine的氨基甲酰基位于活性部位的丝氨酸附近,与丝氨酸发生共价结合后释放出一个酚类化合物(图10)。

图10 乙酰胆碱酯酶的共价抑制剂
组织蛋白酶的共价抑制剂
组织蛋白酶是一组半胱氨酸蛋白酶,参与溶酶体的蛋白水解,并控制多种细胞信号通路。在人类中编码有11种组织蛋白酶(B, C, F, H,K, L, O, S, V, W,和X)这些酶的过度活跃通常与疾病发展有关。
在不同的组织蛋白酶中,CatK一直备受关注。在破骨细胞中含量丰富,在骨吸收和骨重塑中起重要作用。因此,它已经成为治疗骨质疏松症的药物靶点。与其他组织蛋白酶相比,Balicatib对CatK有很大的选择性,但因为导致一些患者出现吗啡样皮肤病变因此在II期临床研究后停用。在许多CatK的共价抑制剂,odonacatib,达到了III期试验(图11b),Odonacatib含有一个丁腈基团,可与CatK to的C25发生反应形成亚氨基硫酯加合物(图11c)。

图11 组织蛋白酶的共价抑制剂
总结与展望
尽管在治疗疾病的历史中,共价抑制剂并不是很受欢迎,除了潜在的毒性和脱靶等缺点外,共价抑制剂发挥作用依赖于靶蛋白或者受体中的单一氨基酸残基,如果此氨基酸发生突变,则会产生耐药性,但共价抑制剂作用较强并且持久,可以降低给药剂量以及给药频率,特别是共价抑制剂有可能作用于传统的不可成药的靶蛋白(例如KRAS),因此共价抑制剂将具有很好的发展前景,期待有更多的共价抑制剂上市。
参考文献:
1、Lagoutte, R., Patouret, R., Winssinger, N., 2017. Covalent inhibitors: an opportunity for rational target selectivity. Current Opinion in Chemical Biology 39, 54–63.
2、Sutanto, F., Konstantinidou, M., Dömling, A., 2020. Covalent inhibitors: a rational approach to drug discovery. RSC Medicinal Chemistry 11, 876–884.
3、Ghosh, A.K., Samanta, I., Mondal, A., Liu, W.R., 2019. Covalent Inhibition in Drug Discovery. ChemMedChem 14, 889–906.

来源:药学速览


