您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-05-07 16:00
摘要 目的:建立反相高效液相色谱法测定苯丙氨酯及其片剂的有关物质和含量。方法:采用Phenomenex Luna C18色谱柱(4.6mm×250 mm,5 μm),以甲醇-水(60∶40)为流动相,流速为1.0mL·min-1,检测波长为259nm,柱温为40℃,进样量为20 μL。结果:有关物质线性范围为 0.51~30.24μg·mL-1,r=0.999 9,检出限为4 ng,精密度、稳定性、重复性试验的RSD≤0.8%;含量测定线性范围为12.60~100.82μg·mL-1,r=0.999 9;苯丙氨酯片的平均回收率为99.7%,RSD=0.6%(n=9);原料含量测定结果为99.3%~99.8%;片剂含量测定结果为99.0%~103.6%。结论:本法简便、准确、专属性好、灵敏度高,可用于苯丙氨酯及其片剂的质量控制。
关键词:苯丙氨酯;高效液相色谱法;有关物质;含量测定;质量控制
苯丙氨酯是一种具有镇静作用的中枢性骨骼肌松弛剂,临床上主要用于腰背、四肢肌腱炎、韧带损伤、肌肉紧张痛、神经痛及风湿性关节炎的治疗[1]。本品治疗效果显著,且无非甾体类抗炎药的严重不良反应,目前国内有多家企业生产,其中原料生产厂家有3家,片剂生产厂家有38家。苯丙氨酯现行标准含量测定方法为氮测定法[2],片剂含量测定方法为紫外分光光度法[2]和氮测定法[3]。采用氮测定法,试验步骤较为繁琐;采用紫外分光光度法,由于采用乙醇为配制溶剂,其挥发性较大,测定结果受实验室温湿度等试验环境的影响较大,易造成测定结果的不准确性。鉴于本品现行标准为《中华人民共和国药典》1995年版,测定方法具有上述缺陷且均无有关物质检查项等问题,国家药典委员会将其列为国家药品标准提高工作,天津市药品检验研究院承担苯丙氨酯及其片剂质量标准的修订起草工作,本文拟建立同时测定苯丙氨酯及片剂含量和有关物质的HPLC方法[4]。
1 材料
LC-20A型高效液相色谱仪①;1200型高效液相色谱仪②;AE-240型电子天平(瑞士MettlerToledo公司);AS 10200A型超声仪(天津奥特赛恩斯仪器有限公司)。
苯丙氨酯对照品;苯丙氨酯(东港市某公司提供,批号:150901、170103、170306);苯丙氨酯片(规格:200mg,由天津某药厂提供,批号:02160208、02160209、P0214039);甲醇为色谱纯,水为纯化水。
2 方法与结果
2.1 色谱条件
色谱柱:①Phenomenex Luna C18柱(4.6 mm×250 mm,5 μm);②Agilent Eclipse XDB-C18柱(4.6 mm×250 mm,5 μm);③Dikma Diamonsil C18柱(4.6 mm×250 mm,5 μm);除片剂有关物质空白辅料影响试验和耐用性试验外,其他试验均采用仪器①和色谱柱①;流动相为甲醇-水(60∶40);检测波长为259 nm;柱温40 ℃;流速1.0 mL·min-1,进样量20 μL。
2.2 溶液的制备
2.2.1 供试品溶液a(有关物质)
取本品原料约50 mg(或相当于苯丙氨酯约50 mg的片粉),精密称定,置50 mL量瓶中,加流动相溶解并稀释至刻度,摇匀(片剂溶液滤过后取续滤液)(1.0 mg·mL-1)。
2.2.2 对照溶液(有关物质)
精密量取“2.2.1”项下供试品溶液a 1 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,即得。
2.2.3 灵敏度测试溶液
精密量取“2.2.2”项下对照溶液1 mL,置20mL量瓶中,用流动相稀释至刻度,摇匀,即得。
2.2.4 空白辅料溶液
按天津某药厂提供的处方比例,照供试品溶液a 配制方法配制不含主成分的辅料溶液。
2.2.5 供试品溶液b(含量测定)
取本品原料约50 mg(或相当于苯丙氨酯约50 mg的片粉),精密称定,置100 mL量瓶中,加流动相溶解并稀释至刻度,摇匀(片剂溶液过滤后取续滤液),精密量取5mL,置50 mL量瓶中,用流动相稀释至刻度,摇匀(50 μg·mL-1)。
2.2.6 对照品溶液(含量测定)
精密称取苯丙氨酯对照品适量,加流动相溶解并定量稀释制成50 μg·mL-1的溶液,摇匀,即得。
2.2.7 专属性试验储备液
精密称取原料药(或片粉)适量,加流动相溶解并定量稀释制成10 mg·mL-1的溶液,摇匀,即得。
2.3 系统适用性试验
取对照品溶液进样测试,按苯丙氨酯峰计算理论板数为14588,拖尾因子为1.1。按“2.1”项下进样、测定、记录色谱图。对照品溶液、供试品溶液a、b及空白溶剂的色谱图见图1,主峰与相邻杂质峰的分离度>1.5。
2.4 专属性试验
2.4.1 破坏试验
取“2.2.7”项下专属性试验储备液各5份,每份1mL,分别进行如下处理。(1)酸破坏:加盐酸溶液1mL,振摇30 min后,用氢氧化钠试液调pH为中性,加流动相稀释至10mL(片粉溶液需过滤,以下同);(2)碱破坏:加氢氧化钠试液1mL,振摇30 min后,用1 mol·L-1盐酸溶液调pH为中性,加流动相稀释至10mL;(3)氧化破坏:加30%过氧化氢溶液1mL,沸水浴中加热15 min,放冷至室温,用流动相稀释至10 mL;(4)光照破坏:光照7 d,加流动相稀释至10mL;(5)热破坏:加流动相适量,沸水浴中加热1 h,放冷至室温,加流动相稀释至10 mL。分别取上述溶液20μL,按“2.1”项下的色谱条件进样、测定,记录色谱图至主成分峰保留时间的4倍,详见图2。结果表明:在上述条件下,原料及片剂供试品溶液在酸、碱、光、热的破坏条件下基本稳定,均未见明显增加的杂质色谱峰,在氧化加热的条件下产生降解杂质,各杂质峰之间、与主峰之间基本可达到基线分离,说明拟定的色谱条件专属性良好。
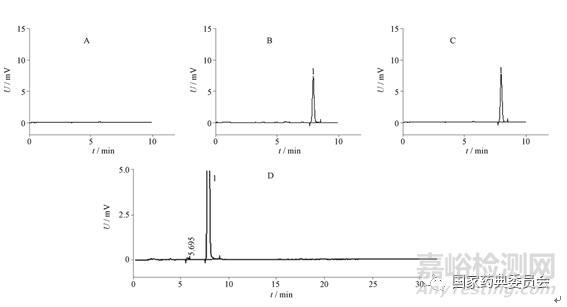
A.空白溶剂(blank solution) B.对照品溶液(reference solution) C.供试品溶液b(sample solution b) D.供试品溶液a(sample solution a) 1.苯丙氨酯(phenprobamate)
图1 高效液相色谱图
2.4.2 辅料干扰试验
取“2.2.4”项下空白辅料溶液20 μL,分别采用以下三种色谱条件,即色谱条件A:仪器①色谱柱①;B:仪器①色谱柱②;C:仪器②色谱柱③,其他按“2.1”项下的色谱条件,进样,记录色谱图,结果显示,在上述色谱条件下,主峰的相对保留时间为0.4之前均产生若干辅料峰,保留时间最大的辅料峰的相对保留时间均为0.4,按自身对照法计算,总辅料量约为0.5%。原料及片剂共6批样品在辅料峰处均未检出杂质色谱峰,且专属性试验的各种破坏条件下亦未在辅料峰处检出明显增加的降解产物峰,故在质量标准中扣除主峰相对保留时间为0.4之前的色谱峰,以去除辅料对片剂有关物质测定的影响。
2.5 线性关系考察
精密称取苯丙氨酯对照品适量,用流动相溶解并稀释制成含苯丙氨酯0.5、1、2、5、10、20的系列浓度溶液(有关物质)和12.5、25、50、75和100μg·mL-1的系列浓度溶液(含量测定),分别取20μL,按“2.1”项下条件进样、测定、记录色谱图。以峰面积(Y)对浓度(X)进行线性回归。(有关物质)和(含量测定)线性方程分别为:
Y=140 1X+39.88 r=0.9999
Y=138 8X+428.3 r=0.9999
结果表明:苯丙氨酯在0.51~30.24μg·mL-1和12.60~100.82 μg·mL-1的浓度范围内与其峰面积均呈良好线性关系。
2.6 检测限与定量限
取苯丙氨酯对照品适量,采用逐步稀释法配制系列浓度的溶液,按“2.1”项下条件进样、测定、记录色谱图。主成分峰信噪比S/N=10时,以此作为灵敏度测试溶液(0.05%),最低定量限为10 ng;信噪比S/N=3时,最低检出限为4 ng。
2.7 精密度试验
取含量测定对照品溶液连续进样6次,苯丙氨酯峰面积的RSD=0.2%,表明本系统精密度良好。
2.8 稳定性试验
取原料供试品溶液a和b,分别于室温下放置0、2、8、12、24 h时进样测定。供试品溶液a苯丙氨酯峰面积的RSD分别为0.4%(原料)和0.3%(片剂);供试品溶液b苯丙氨酯峰面积的RSD分别为0.8%(原料)和0.5%(片剂)。结果表明有关物质及含量测定供试品溶液均在24 h内基本稳定。
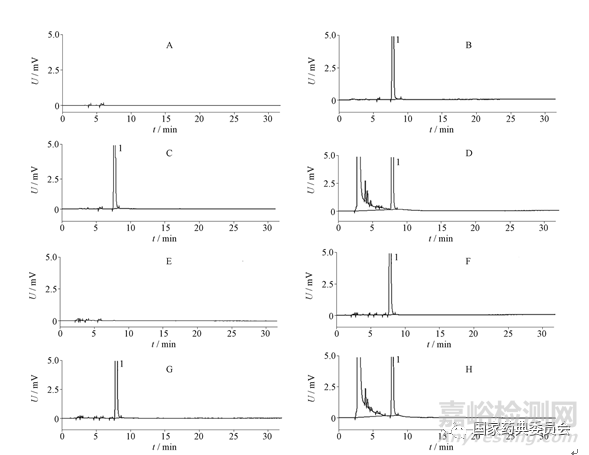
A.空白溶剂(blank solution) B.原料供试液(phenprobamate solution) C.原料酸/碱/高温/光照破坏(phenprobamate destroyed by acid/alkali/heat/light) D.原料氧化破坏(phenprobamate destroyed by oxidation) E.片剂辅料溶液(excipient solution)F.片剂供试液(phenprobamate tablets test solution) G.片剂酸/碱/高温/光照破坏(phenprobamate tablets destroyed by acid/alkali/heat/light) H.片剂氧化破坏(phenprobamate tablets destroyed by oxidation) 1.苯丙氨酯(phenprobamate)
图2 专属性试验高效液相色谱图
2.9 重复性试验
取苯丙氨酯原料,分别按“2.2.1”和“2.2.5”制备供试品溶液a和b各6份,分别按“2.1”项下条件进样、测定、记录色谱图。检出有关物质单个最大杂质和杂质总量结果基本一致;含量测定结果RSD=0.2%,表明方法重复性好。
2.10 片剂回收率试验
称取空白全辅料约0.25片量,共9份,分别置100 mL量瓶中,精密加入对照品约40、50和60 mg各3份,加流动相适量,振摇使溶解后用流动相稀释至刻度,摇匀,滤过,精密量取续滤液5 mL,置50 mL量瓶中,用流动相稀释至刻度,摇匀,分别按“2.1”项下条件进样、测定、记录色谱图。计算平均回收率为99.7%,RSD=0.6%(n=9),表明方法准确度良好。
2.11 耐用性试验
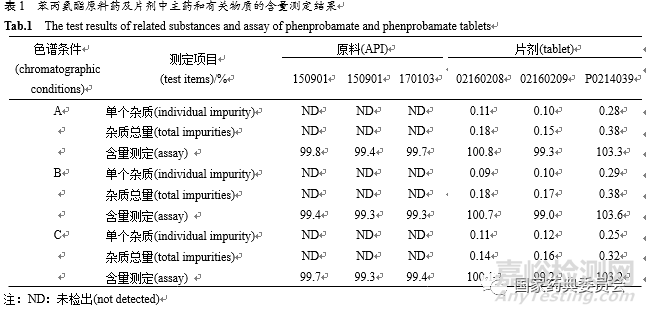
按“2.4.2”项下的三种色谱条件,分别对苯丙氨酯原料及片剂进行有关物质和含量的测定,测定结果基本一致(见表1),表明本方法的耐用性良好。
2.12 样品测定
2.12.1 主药含量测定
取原料药及片剂各3批,按“2.2.5”项和“2.2.6”项下方法制备,取供试品溶液和对照品溶液各20 μL注入液相色谱仪,记录色谱图,按外标法计算含量,结果见表1。
2.12.2 有关物质测定
取原料药及片剂各3批,按“2.2.1”项下方法制备,取供试品溶液和对照溶液各20 μL注入液相色谱仪,记录色谱图至主成分峰保留时间的2倍,按主成分自身对照法计算杂质含量,结果见表1。
3 讨论
3.1 供试液浓度的选择[5]
以甲醇-水(60∶40)为流动相,首先按含量测定供试液浓度为0.5 mg·mL-1、有关物质供试液浓度为2.5 mg·mL-1或5 mg·mL-1、以1%的稀释溶液为自身对照进行考察,采用仪器①和上述3支色谱柱。结果显示,高浓度供试液(2.5 mg·mL-1和5 mg·mL-1)色谱图中,主成分色谱峰的峰形不对称、拖尾严重,拖尾因子在3支色谱柱上均为1.3~1.4。经调节流动相组成及比例亦不能有效改善该色谱峰的对称性。将有关物质供试液浓度调整为1.0 mg·mL-1,含量测定供试液浓度调整为50 μg·mL-1,以10 μg·mL-1的溶液作为1%的自身对照溶液,上述浓度的溶液在3支色谱柱上均峰形对称性良好(拖尾因子≤1.2),以0.5μg·mL-1的溶液作为灵敏度测试溶液(相当于0.05%),主成分峰信噪比均>10,可以满足测定要求。
3.2 检测波长的选择
取浓度为1.0 mg·mL-1的供试液在200~400nm波长范围内扫紫外图谱,显示本品的最大吸收波长为259 nm;在上述液相色谱条件下苯丙氨酯的最大吸收波长亦为259 nm,故确定以259 nm作为测定波长。
3.3 测定时间的选择
根据专属性试验的色谱图及6批样品有关物质的测定图谱,在记录色谱图至主成分峰保留时间的2倍~4倍时,均未检出杂质色谱峰,故确定色谱图的记录时间为主成分峰保留时间的2倍。
3.4 与原标准测定方法的比较
本研究建立的RP-HPLC法较氮测定法操作更为简便;较紫外分光光度法可避免挥发性溶剂所带来的误差,使测定结果更为准确。另外,现行标准均未控制杂质含量,难以保证用药安全,而本研究建立的有关物质测定方法灵敏度高,可以准确测定杂质含量。
3.5 小结
本方法简便、准确,专属性好,灵敏度高,可用于苯丙氨酯及其片剂的质量控制。
参考文献
[1] 苏聪娟,黄占周,于林怡,等.苯丙氨酯药理毒理和临床研究综述. 临床合理用药,2012,5(12A):146
SU CJ, HUANG ZZ, YULY, et al. A review of pharmacotoxicology and clinical studies of phenylalanine. Chin J Clin Ration Drug Use,2012,5(12A):146
[2] 中华人民共和国药典1995年版.二部. 1995:360
ChP1995.VolⅡ. 1995:360
[3] 国家药品标准·化学药品地方标准上升国家标准.第十四册. 2003:32
National Drug Standards: National Standards for Chemicals Promoted from Local Standards.Vol 14. 2003:32
[4] 谢沐风.如何建立高效液相色谱法测定有关物质的方法. 中国医药工业杂志,2007,38(1):45
XIE MF.How to establish a HPLC method for determination of related substances in drugs. Chin J Pharm,2007,38(1):45
[5] 谢沐风.高效液相色谱法测定含量时关于确立色谱条件与溶液浓度的讨论. 中国药品标准,2008,9(4):288
XIE MF. Discussion about chromatographic conditions and solution concentration in content determination by HPLC. Drug Stand China,2008,9(4):288

来源:中国药品标准


