您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-06-28 21:26
以紫杉醇及紫杉醇注射液为例,根据《化学药物杂质研究的技术指导原则》,并参考《中国药典》《美国药典》《欧洲药典》《英国药典》,探讨单一成分药物有关物质质量控制研究要点:(1)不同来源(例如天然来源分离、半合成、发酵)单一成分药物的有关物质可能存在差异;(2)不同来源单一成分新药原料药与制剂的有关物质控制侧重点有所不同,新药制剂的有关物质侧重于对降解产物制定限度;(3)4国药典收录的紫杉醇及其注射剂在有关物质的控制上有所区别;(4)4国药典或相关指导原则对有关物质限度的要求基本一致;(5)通过中药注册分类途径申报的,从天然来源分离得到的单一成分药物及制剂需参照中国《化学药物杂质研究的技术指导原则》的相关要求进行研究和控制。
单一成分药物,即药物的活性成分是单个化合物,可能来源于不同的制备工艺——天然来源分离制备、半合成制备、发酵制备。有机杂质的化学结构一般与活性成分类似或具渊源关系,故通常又可称之为有关物质。不同的制备工艺可能产生不同的有关物质种类及数量。天然来源分离得到的单一成分药物的申报途径可以是化学药或中药。基于“以患者为中心”的药物研发理念,需坚守底线,确保药物的安全性,无论按哪一种途径申报,对天然来源分离得到的单一成分药物有关物质质量控制的研究关注点应保持一致。
本文就来源于不同制备工艺(天然来源分离、半合成、发酵)的单一成分药物——紫杉醇及其注射剂的《中国药典》《美国药典》《欧洲药典》《英国药典》4国药典质量标准进行对比分析,旨在了解:(1)不同来源单一成分药物的有关物质是否存在差异;(2)不同来源单一成分药物与制剂的有关物质控制的异同点;(3)4国药典收录的紫杉醇及其注射剂在有关物质控制上的区别;(4)4国药典或指导原则对有关物质限度要求的区别;(5)天然来源分离制备所得单一成分药物的有关物质研究要求。
1、有关物质的相关概念
不同来源的单一成分药物的杂质(impurity)中,一般包含一些有机杂质,又称为有关物质(relatedsubstances)。药物的有关物质中,与药物共存的异构体和抗生素多组分可称为共存物质或伴随成分(concomitant components)。从杂质的安全性角度进行划分,无明显不良生物活性的物质可称为普通杂质(ordinary impurities),具有明显不良生物活性的物质称为有毒杂质。
1.1有关物质
中国《化学药物杂质研究的技术指导原则》[1]对有关物质、有机杂质的概念进行了解释。有机杂质包括工艺中引入的杂质和降解产物等,可能是已知的或未知的、挥发性的或不挥发性的。由于这类杂质的化学结构一般与活性成分类似或具渊源关系,故通常又可称之为有关物质。
1.2杂质
《美国药典》(USP)43-NF38“1086原料药和药品中的杂质”[2]对杂质的概念进行了解释。杂质是指原料药中非原料药化学实体的任何成分和(或)制剂中非配方成分的任何成分。
按杂质的理化性质划分,可分为有机杂质、无机杂质、残留溶剂等,可以理解为杂质的概念中包含有关物质。
1.3普通杂质
USP43-NF38“466普通杂质”[3]对普通杂质的概念进行了解释。普通杂质是指原料药和(或)制剂中无明显不良生物活性的物质。除非药物专论中另有规定,否则普通杂质总量不得超过2.0%。如果某一药物专论对伴随成分和(或)特定杂质或降解产物设定了限制,则这些成分不包括在普通杂质中。
根据USP43-NF38对普通杂质的定义可知,普通杂质的总量一般不得超过2.0%,且可根据药物杂质的具体情况再进行调整。
1.4共存物质或伴随成分
《中国药典》2020年版四部“9102药品杂质分析指导原则”[4]对共存物质的概念进行了解释。共存的异构体和抗生素多组分一般不作为杂质检查项目,作为共存物质,必要时在质量标准中规定其比例,以保证生产用的原料药与申报注册时的一致性。但当共存物质为毒性杂质时,该物质就不再认为是共存物质。
USP43-NF38“466普通杂质”对伴随成分的概念进行了解释。伴随成分是指原料药的特征性成分,这些成分通常不被视为杂质。伴随成分的例子是立体和旋光异构体(或外消旋物)和抗生素的混合物。具有明显不良生物活性的有毒杂质不被视为伴随成分。
由上可知,共存物质又可称为伴随成分,其对应的概念较小,例如药物共存的异构体和抗生素多组分。值得关注的是,对于天然来源分离制备所得单一成分药物,其有关物质中未必含有共存物质/伴随成分,需加强对有关物质的研究控制。
2、药物有关物质研究要点
根据中国《化学药物杂质研究的技术指导原则》[1],建议关注以下3个方面。
2.1药物有关物质研究的基本原则
在药物有关物质的研究中,应遵循的基本原则:按《化学药物杂质研究的技术指导原则》要求控制每个已知杂质、未知杂质及总杂质的限度。共存的异构体和抗生素的多组分一般不作为杂质进行控制,必要时作为共存物质在质量标准中规定其比例。单一的对映体药物,其对映异构体应作为杂质控制。允许含一定量无害或低毒的共存物,但对有毒杂质则应严格控制。毒性杂质的确认主要依据安全性试验资料或文献资料。尽管直接用分离纯化的杂质进行安全性研究比较合适,但也可采用含有杂质的原料药进行研究。
2.2创新药物有关物质的限度制定具一定灵活性
由于在创新药物的研究过程中,需通过一系列的药理毒理及临床研究来验证该药品的安全有效性,而研究所用的样品本身会包含一定种类与数量的杂质,所以如果在这些研究中并未明显反映出与杂质有关的毒副作用,即使有些杂质的含量超出了《化学药物杂质研究的技术指导原则》附件1、2的质控限度,仍可认为该杂质的含量已经通过了安全性的验证。在此前提之下,如果该杂质的含量同时也在正常的制备工艺所允许的限度范围内,那么根据试验样品中杂质的含量所确定的限度可认为是合理的。在特殊情况下,应具体问题具体分析,在保证安全的前提下,可以与《化学药物杂质研究的技术指导原则》附件1、2中杂质的限度不一致,并同时提供修改限度的充分理由。
2.3新药制剂中有关物质的研究思路
除降解产物和毒性杂质外,已在原料药质量标准中控制,且在制剂过程中含量没有增加的杂质,制剂中一般不再控制。
3、4国药典收录的紫杉醇质量标准对比
在《中国药典》2020年版[5]、《美国药典》USP43-NF38[6]、《欧洲药典》10.2[7]、《英国药典》BP2020[8]中,均收录了紫杉醇原料药,本文就以紫杉醇原料药为例,比较4国药典中有关物质质量控制的要求。
3.1、4国药典收录的紫杉醇质量标准检测项比较
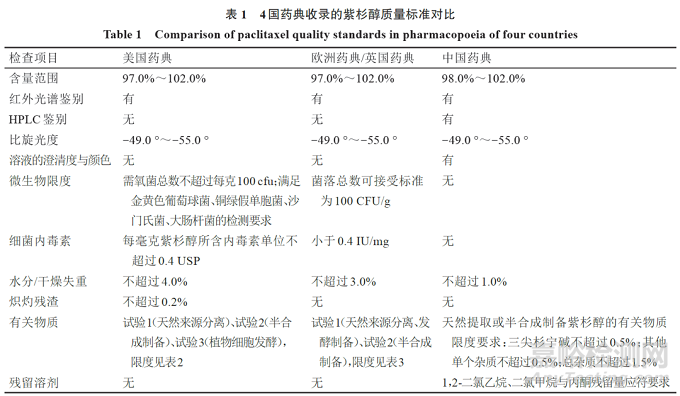
将4国药典收录的紫杉醇质量标准检测项进行比较,见表1。可知:(1)《中国药典》紫杉醇的含量限度较美国、欧洲、英国药典范围更窄,水分或干燥失重的限度更低;(2)《中国药典》紫杉醇质量标准包含HPLC鉴别、溶液的澄清度与颜色、残留溶剂检测项,而这3项均未列入美、欧、英药典标准;(3)微生物限度、细菌内毒素、炽灼残渣检测项未列入我国药典紫杉醇质量标准。
3.2《美国药典》收录的紫杉醇有关物质限度
《美国药典》收录的紫杉醇有关物质限度可知,对于3种来源的紫杉醇,有对应的3套有关物质限度:(1)天然来源分离制备的紫杉醇,已鉴定杂质为10个,每个杂质的限度最多不超过0.5%;(2)半合成制备的紫杉醇,已鉴定杂质为13个,每个杂质的限度最多不超过0.7%;(3)植物细胞发酵制备的紫杉醇,已鉴定杂质为8个,每个杂质的限度最多不超过0.5%。见表2。
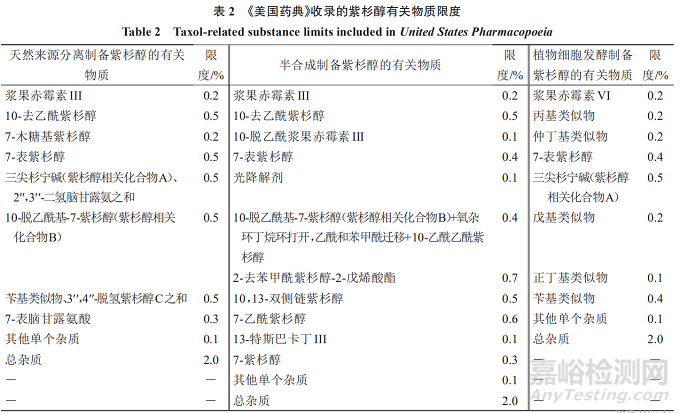
其中天然来源分离和半合成制备的紫杉醇有关物质中,有4个杂质(浆果赤霉素III、10-去乙酰紫杉醇、7-表紫杉醇、10-脱乙酰基-7-紫杉醇)成分一致,限度略有区别;天然来源分离和发酵制备的紫杉醇有关物质中,有2个杂质(7-表紫杉醇、三尖杉宁碱)成分一致,限度略有区别;3种来源的紫杉醇有关物质中,有1个杂质(7-表紫杉醇)成分一致,限度略有区别。
《美国药典》收录的3种来源的紫杉醇有关物质中,其他单个杂质(未鉴定杂质)限度均为0.1%,总杂质限度均为2.0%,且对于超过0.1%的单个杂质均进行了鉴定。3种来源的紫杉醇在有关物质的控制上有一定区别,3种制备工艺所得的相同的有关物质较少,提示需注意不同来源单一成分药物的有关物质研究,按《化学药物杂质研究的技术指导原则》的质控要求,对已鉴定和未鉴定有关物质制定限度。
3.3《欧洲药典》/《英国药典》收录紫杉醇有关物质限度
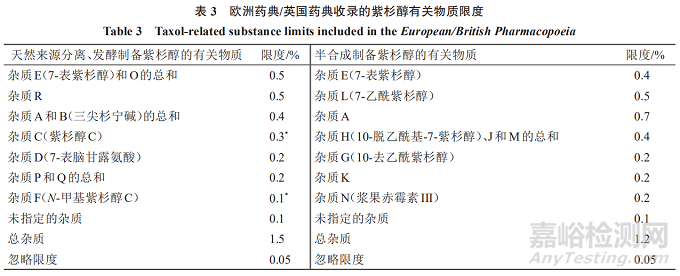
《欧洲药典》/《英国药典》收录的紫杉醇有关物质限度可知,对于3种来源的紫杉醇有对应的2套有关物质限度:(1)天然来源分离制备及发酵制备的紫杉醇,已鉴定杂质为10个,每个杂质的限度最多不超过0.5%;(2)半合成制备的紫杉醇,已鉴定杂质为9个,每个杂质的限度最多不超过0.7%。见表3[标*者为对照溶液(d)获得的色谱图中主峰面积的比值,未标*的均为对照溶液(a)获得的色谱图中主峰面积的比值]。
欧洲药典/英国药典收录的3种来源的紫杉醇有关物质中,有2个杂质(7-表紫杉醇、杂质A)成分一致,限度略有区别。其他单个杂质(未鉴定杂质)限度均为0.1%,总杂质限度为1.5%、1.2%,忽略限度均为0.05%,且对于超过0.1%的单个杂质均进行了鉴定。3种来源的紫杉醇在有关物质的控制上有一定区别,天然来源分离和发酵制备所得紫杉醇的有关物质限度一致,而半合成制备所得紫杉醇的有关物质限度则单独制定。
3.4《中国药典》收录的紫杉醇有关物质限度
《中国药典》收录的紫杉醇来源有2种,即天然提取或半合成制备。对于这2种来源的紫杉醇,有对应的一套有关物质限度:三尖杉宁碱不得超过0.5%;其他单个杂质不得过0.5%;总杂质不得超过1.5%。已鉴定并制定限度的杂质为1个(三尖杉宁碱),另有限度大于0.1%的单个杂质未进行鉴定,但控制了未鉴定的杂质限度为0.5%。
《中国药典》收录的紫杉醇来源有2种,即天然提取或半合成制备。对于这2种来源的紫杉醇,有对应的一套有关物质限度:三尖杉宁碱不得超过0.5%;其他单个杂质不得过0.5%;总杂质不得超过1.5%。已鉴定并制定限度的杂质为1个(三尖杉宁碱),另有限度大于0.1%的单个杂质未进行鉴定,但控制了未鉴定的杂质限度为0.5%。
4、《中国药典》《美国药典》收录的紫杉醇注射液质量标准对比
在4国中,紫杉醇注射液仅收录于《中国药典》[5]、《美国药典》[9]。
4.1紫杉醇注射液质量标准检测项比较
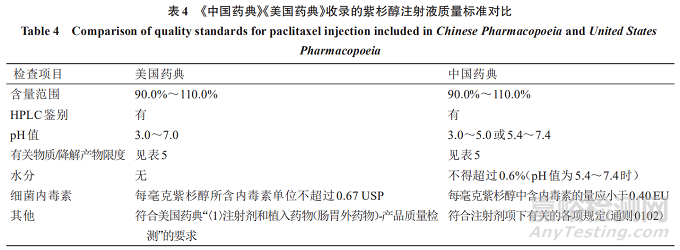
将《中国药典》《美国药典》收录的紫杉醇注射液质量标准检测项进行比较,可知(1)《中国药典》紫杉醇注射液的pH值范围限定更精确;(2)《中国药典》紫杉醇注射液质量标准有水分检测项,而该项未列入美国药典的紫杉醇注射液标准。见表4。
4.2《中国药典》《美国药典》收录的紫杉醇注射液有关物质限度比较
《美国药典》收录的紫杉醇注射液降解产物限度可知,对于3种来源的紫杉醇原料药,其制剂共用一套降解产物限度:已鉴定杂质为5个,每个杂质的限度最多不超过0.8%。其中4个杂质(浆果赤霉素III、10-去乙酰紫杉醇、7-表紫杉醇、10-脱乙酰基-7-紫杉醇)为天然来源分离和半合成制备的紫杉醇原料药有关物质成分,4个成分在紫杉醇注射液中的限度略高于在紫杉醇原料药中的限度。其他单个降解产物(未鉴定杂质)限度均为0.1%,总杂质限度为2.0%,且对于超过0.1%的单个降解产物均进行了鉴定。见表5。
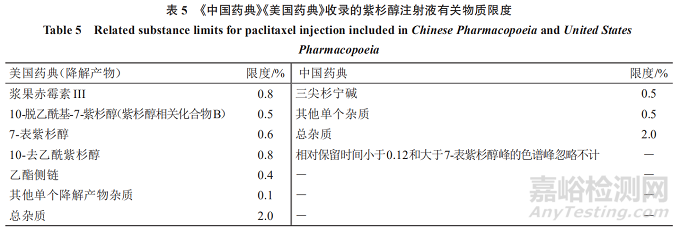
《中国药典》收录的紫杉醇注射液有关物质限度可知,对于2种来源的紫杉醇原料药,其制剂共用一套降解产物限度:三尖杉宁碱不得超过0.5%;其他单个杂质不得超过0.5%;总杂质不得过2.0%。已鉴定并控制的杂质为1个(三尖杉宁碱),另有限度大于0.1%的单个杂质未进行鉴定,但控制未鉴定的杂质限度为0.5%。与紫杉醇原料药的有关物质限度相比,紫杉醇注射液比紫杉醇原料药总杂质限度有所提高,其余未变。见表5。
《美国药典》收录的紫杉醇注射液有关物质限度显示,主要是针对紫杉醇的降解产物进行研究,而紫杉醇原料药的有关物质中已经控制了的非降解产物杂质未重复列入紫杉醇注射液有关物质检测项目。中国药典收录的紫杉醇注射液有关物质研究存在与紫杉醇原料药有关物质研究类似的问题,未对含量较高的杂质进行鉴定。建议关注新药制剂有关物质研究的基本思路:除降解产物和毒性杂质外,已在原料药质量标准中控制,且在制剂过程中含量没有增加的杂质,制剂中一般不再控制。
5、4国药典或相关指导原则对药物有关物质的研究要求比较
中国《化学药物杂质研究的技术指导原则》中新药的原料药或制剂有关物质的报告限度、鉴定限度、质控限度要求均与人用药品注册技术国际协调会议(ICH)《Q3A(R2)新原料药杂质》[10]、《Q3B(R2)新药产品中的杂质》[11]保持一致。在上述指导原则中未对新药原料药或制剂的总杂质制定限度。《美国药典》限定了普通杂质的总量一般不得超过2.0%,且可根据药物杂质的具体情况再进行调整。
值得注意的是,ICH指导原则通过制定制剂中降解产物的限度对制剂中的有关物质进行控制,美国药典中紫杉醇注射液也是对降解产物制定限度。提示在对制剂的有关物质研究中,可考虑制剂有关物质的控制侧重点与原料药不同,重点在于对降解产物的控制;另外,除降解产物和毒性杂质外,已在原料药质量标准中控制,且在制剂过程中含量没有增加的杂质,制剂中一般不再控制。
6、结语
《中共中央关于制定国民经济和社会发展第十四个五年规划和二〇三五年远景目标的建议》[12]指出:“保障人民生命安全”“强化生物安全保护,提高食品药品等关系人民健康产品和服务的安全保障水平”。《国家药监局关于促进中药传承创新发展的实施意见》[13]指出:“坚守底线,强化中药质量安全监管”“加强中药安全性研究”“建立符合中药特点的安全性评价方法和标准体系,建立以中医临床为导向的中药安全性分类分级评价策略”。药品审评工作中,需树立底线思维,坚持人民至上、生命至上,把保护人民生命安全摆在首位。对于现阶段科学技术水平可以达到的领域,鼓励应用新技术、新方法、新工具进行药品安全的基础研究。
本文通过分析4国药典收录的药物实例——紫杉醇及其注射剂质量标准,结合4国药典及相关指导原则对于有关物质的研究要求,可总结出以下5点:
(1)不同来源单一成分药物的有关物质可能存在差异,对不同来源(天然来源分离、半合成、发酵)单一成分药物的有关物质建议进行充分的研究,并符合相关要求。
(2)不同来源单一成分新药原料药与制剂的有关物质控制侧重点有所不同,新药制剂的有关物质侧重于对降解产物制定限度。另外,除降解产物和毒性杂质外,已在原料药质量标准中控制,且在制剂过程中含量没有增加的杂质,制剂中一般不再控制。
(3)4国药典收录的紫杉醇及其注射剂在有关物质的控制上有所区别,美国、欧洲、英国药典对于不同来源的紫杉醇有关物质控制体现了不同来源原料药所对应的有关物质的差异。(4)4国药典或指导原则对新药原料药及制剂有关物质的报告限度、鉴定限度、质控限度要求基本一致。除了美国药典对普通杂质的总量进行了限定以外,在相关指导原则中尚未对新药原料药或制剂的总杂质制定限度。
(5)对于天然来源分离制备所得单一成分药物,其有关物质中未必含有共存物质/伴随成分,需加强对有关物质的研究控制。通过中药注册分类途径申报的,从天然来源分离得到的单一成分药物及制剂,在目前尚无对应的有效成分中药杂质研究相关技术要求文件的前提下,需参照中国《化学药物杂质研究的技术指导原则》中新药的原料药或制剂有关物质的相关要求进行研究,并结合研究的实际情况制定合理的杂质限度。
作者:孙昱1,郑蕊2*,文海若3*
1.国家药品监督管理局药品审评中心,北京100022
2.中国中医科学院西苑医院,北京100091
3.中国食品药品检定研究院,北京100050

来源:Internet


