您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-07-04 22:58
分子诊断技术是用分子生物学方法针对人体及各种病原体的遗传物质的表达及结构进行检测,从而达到预测及诊断疾病的目的。
近年来,随着分子诊断技术的升级迭代,分子诊断的临床应用越来越广泛和深入,分子诊断市场进入快速发展期。
笔者总结市场上常见的分子诊断技术,分为上中下三篇,上篇介绍PCR技术,中篇介绍核酸等温扩增技术,下篇介绍测序技术。
01、上篇:PCR技术
PCR技术
PCR (polymerase chain reaction)聚合酶链式反应,是体外DNA扩增技术之一,至今已经超过30年的历史。
PCR技术于1983年由美国Cetus公司的KaryMullis首创,1985 年Mullis申请了PCR专利,同年在Science上发表了第一篇PCR 学术论文,1993 年Mullis因此获得了诺贝尔化学奖。
PCR基本原理
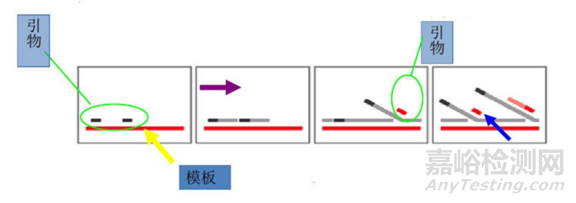
PCR可以将目标DNA片段扩增一百万倍以上,其原理是在DNA聚合酶催化下,以母链DNA为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。

标准 PCR 过程分为三步:
1.变性(Denaturation):利用高温使DNA 双链分离。DNA双链之间的氢键在高温下(93- 98℃)被打断。
2.退火(Annealing):在 DNA 双链分离后,降低温度使得引物可以结合于单链DNA 上。
3.延伸(Extension):DNA 聚合酶由降温时结合上的引物处开始沿着DNA 链合成互补链,延伸完成,则完成一轮循环,DNA片段数增加一倍。
往复循环这三个步骤25-35 次,DNA 片段数将得到指数级增加。
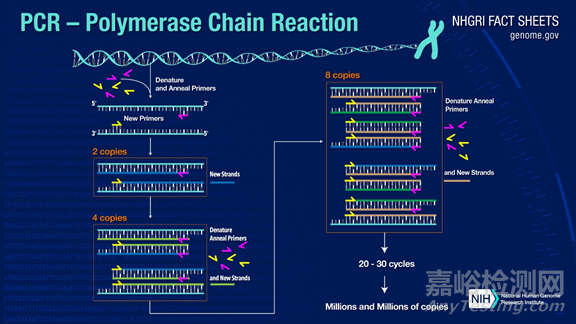
PCR的巧妙之处在于针对不同的目标基因可以设计不同的引物,使目标基因片段在短时间内得到百万级的放大。
目前为止,PCR可以分为三类,分别是普通PCR、荧光定量PCR和数字PCR。
第一代普通PCR
采用普通PCR 扩增仪来对靶基因进行扩增,然后采用琼脂糖凝胶电泳对产物进行检测,只能做定性分析。
第一代PCR主要缺点:
容易发生非特异性扩增和假阳性结果。
检测耗时长,操作繁琐。
只能做定性检测。
第二代荧光定量PCR
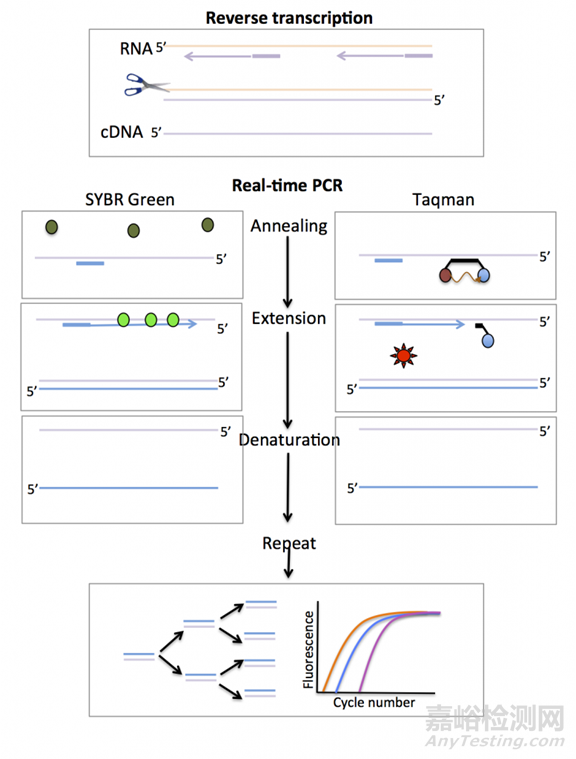
荧光定量PCR(Real-Time PCR),也叫做qPCR,通过在反应体系中加入能够指示反映进程的荧光探针,通过荧光信号的积累来监测扩增产物的积累,通过荧光曲线来判断结果,并可以借助Cq 值和标准曲线来定量。
qPCR 技术由于操作过程在封闭体系中进行,降低了污染概率,并且可以通过对荧光信号监测从而进行定量检测,因此临床应用最为广泛,已成为PCR中的主导技术。
实时荧光定量PCR所使用的荧光物质可分为:TaqMan荧光探针、分子信标和荧光染料。
1)TaqMan荧光探针:
PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。
探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5'-3'外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。
2)SYBR荧光染料:
在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料非特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。SYBR仅与双链DNA进行结合,因此可以通过溶解曲线,确定PCR反应是否特异。
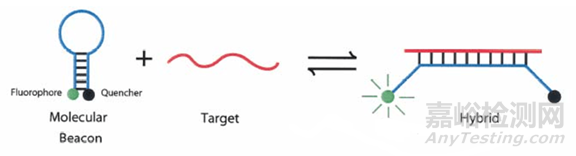
3)分子信标
是一种在5和3末端自身形成一个8个碱基左右的发夹结构的茎环双标记寡核苷酸探针,两端的核酸序列互补配对,导致荧光基团与淬灭基团紧紧靠近,不会产生荧光。

PCR产物生成后,退火过程中,分子信标中间部分与特定DNA序列配对,荧光基因与淬灭基因分离产生荧光。
第二代PCR主要缺点:
灵敏度还有欠缺,低拷贝标本检测不准确。
存在背景值影响,结果易受干扰。
当反应体系中有PCR抑制物时,检测结果易受干扰。
第三代数字PCR
数字PCR(DigitalPCR, dPCR, Dig-PCR)通过终点检测计算目标序列的拷贝数,无需采用内参和标准曲线即可进行精确的绝对定量检测。
数字PCR采用终点检测,不依赖于Ct值(循环阈值),所以数字PCR反应受扩增效率的影响降低,对PCR反应抑制物的耐受能力提高,具有很高的准确度和重现性。
由于具备高灵敏度、高精确度的特点,不易被PCR反应抑制剂干扰,无需标准品可实现真正意义的绝对定量,而成为研究和应用热点。
根据反应单元的不同形式,主要可分为微流体式、芯片式和微滴式三大类系统。
1)微流体数字PCR,Microfluidic digital PCR,mdPCR。
基于微流控技术,对DNA模板进行分液,微流控技术能实现样品纳升级或更小液滴的生成,但液滴需要特殊吸附方式再与PCR反应体系结合,mdPCR已逐渐被其他方式取代。
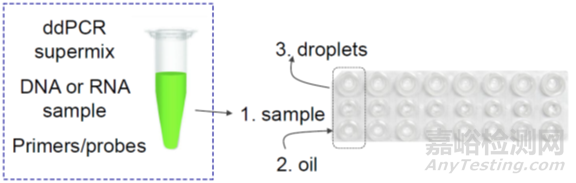
2)微滴数字PCR,Droplet-based digital PCR,ddPCR。
利用油包水微滴生成技术对样品进行微滴化处理,将含有核酸分子的反应体系分成成千上万个纳升级的微滴,其中每个微滴或不含待检核酸靶分子,或者含有一个至数个待检核酸靶分子。
以伯乐的ddPCR为例,主要包括三个步骤:

第一,准备样本和生成微滴,如下图,8x3的微孔板,一排加样本,一排加油滴,通过微滴生成器每个样本生成20000个微滴。

第二,进行“油包水”PCR,把微孔板放入PCR扩增仪,对每个微滴进行40个热循环的PCR反应。
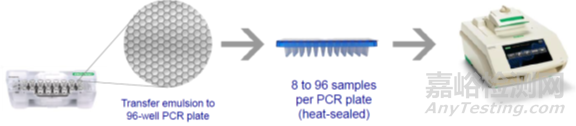
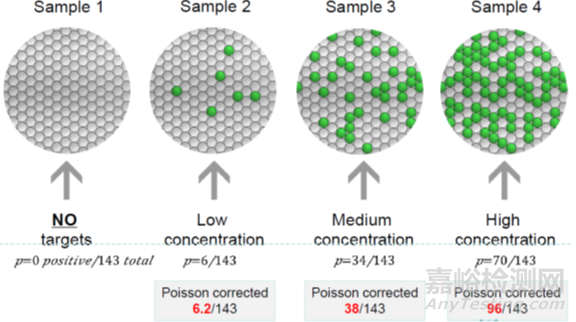
第三,读取微滴结果,把微孔板放入读取仪,通过流式细胞技术获取每个微滴PCR终点结果的荧光信号,并用泊松分布原理纠正结果。
Bio-rad去年上市的Bio-Rad QX ONE数字PCR系统整合了微滴生成、热循环反应及微滴读取等步骤,最大程度减少人工操作。
配备四个独立的荧光检测通道,结合ddPCR特有的高阶多重PCR技术,可以在单反应孔中实现8重拷贝数变异(CNV)检测、5重突变检测或4重基因表达检测。

3)芯片数字PCR,Chip-baseddigital PCR,cdPCR。
利用集成流体通路技术在硅片或石英玻璃上刻上许多微管和微腔体,通过不同的控制阀门控制溶液在其中的流动,将样本液体分成大小一致的纳升级于反应孔种进行数字PCR反应,实现绝对定量。
以法国Stilla technologies 公司的cdPCR技术为例,主要包括以下步骤:
第一,加样和生成微滴:将样本和PCR反应液加入微流控芯片,放入仪器中,仪器可通过物理方法生成单层平铺的20000~30000个微滴阵列。
第二,对每个微滴进行PCR扩增:在Naica Geode微滴生成扩增系统自动进行PCR扩增。
第三,读取和分析结果:将芯片置于Prism微滴读取分析系统上进行荧光信号采集,计数阴阳性微滴,通过泊松分布计算获得靶标基因绝对拷贝数浓度。
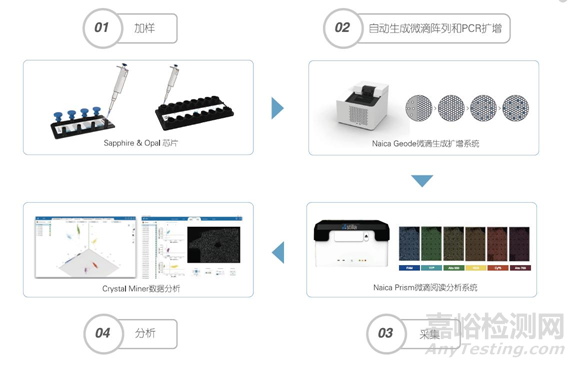
总结一下,将样本和PCR反应液加入微流控芯片,放入仪器中,通过物理方法生成单层平铺的微滴阵列,随后对每个微滴进行扩增,然后进行六通道荧光信号采集,计数阴阳性微滴,通过泊松分布计算获得靶标基因绝对拷贝数浓度。
第三代PCR主要缺点:
仪器和试剂昂贵。
模板质量要求较高,模板量超过微体系量将导致无法定量,过少则定量准确度降低。
当存在非特异性扩增时也会产生假阳性。
PCR的各种延伸技术
1.递减PCR(touchdown PCR):前几循环温度逐渐下降。
2.逆转录PCR(RT-PCR):以由mRNA逆转录而来的cDNA 为模板,也因为是从表现型基因来进行增量的,由此产生出来的cDNA 产物不带有内含子(基因中不具意义的段落),常应用于分子克隆技术。
3.热启动PCR(hotstart PCR):以高热激活型核酸聚合酶进行反应,减少非专一性产物。
4.巢式PCR(nested PCR):先用低特异性引物扩增几个循环以增加模板数量,再用高特异性引物扩增。
5.多重PCR(multiplex PCR):在同一个管中使用多组引物。
6.复原条件PCR(reconditioning PCR):PCR 产物稀释 10 倍后重新放入原浓度的引物和dNTP 等循环 3 次,以消除产物中的异二聚体。
7.dsRNA 合成(dsRNA replicator):合并使用high-fidelity DNA polymersae、T7RNA聚合酶与Phi6 RNA replicase;从双股 DNA转录为对应的双股RNA(dsRNA)。可应用于RNAi实验操作。
8.COLD-PCR (co-amplification at lower denaturation temperature PCR):用以检测突变或特殊等位基因的PCR 应用技术。
参考文献:
1.刘婉彤等,分子诊断技术的临床应用进展
2.杨柳等,非鳞非小细胞肺癌基因检测技术研究进展
3.G.Terrance Walker,etc.Strand displacement amplification-an isothermal, in vitro DNA amplification technique
3.各公司官网
02、中篇:核酸等温扩增技术
本篇介绍一下等温扩增技术。
PCR是使用最为广泛的核酸扩增技术,以其灵敏性、特异性得到广泛应用,然而PCR需要反复的热变性,无法摆脱依赖仪器设备的局限,从而限制了其在临床现场检测中的应用。
自20世纪90年代初,很多实验室开始发展无需热变性的恒温扩增技术,现已开发出环介导等温扩增技术、链替代等温扩增技术、滚环等温扩增技术、依赖核酸序列等温扩增技术等技术。
环介导等温扩增
环介导等温扩增(loop-mediated isothermal amplification,LAMP)是由日本荣研公司的Notomi等于2000年首先提出来的新的核酸扩增技术。
基于该技术,荣研还曾和国内某公司引起专利纠纷。
其扩增原理是基于DNA在65℃左右处于动态平衡状态,任何一个引物向双链DNA的互补部位进行碱基配对延伸时,另一条链就会解离,变成单链。
DNA在此温度下利用4条特异性引物依靠一种链置换DNA聚合酶,使链置换DNA的合成不停地自我循环。
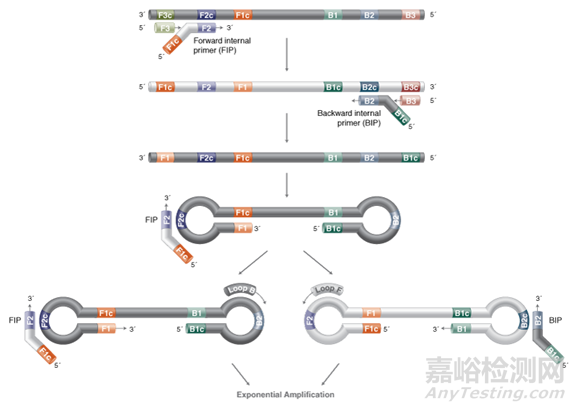
先确定目标基因上的6个特异性区域F3、F2、F1、B1、B2、B3,然后依据这6 个特异性区域设计4条引物(如下图):
正向内引物(forwardinner primer,FIP),由F1c 和F2 组成。
反向内引物(backwardinner primer,BIP),由B1c 和B2 组成,中间均以TTTT作为间隔。
外引物F3、B3 分别由目的基因上的F3、B3 区域组成。
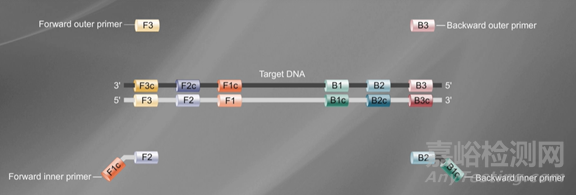
在LAMP 反应体系中,内引物的浓度几倍于外引物的浓度。内引物先与模板链结合合成互补链,形成DNA双链。随后外引物再与该模板链结合,形成DNA双链,在BstDNA 聚合酶的作用下,释放出由内引物合成的互补链,该互补链经过一系列反应最终形成具有哑铃结构的DNA单链。
以哑铃结构DNA单链自身为模板不断形成一端开口的过渡性茎环结构DNA,由内外引物引导过渡性茎环结构DNA不断发生链置换延伸反应,最后形成具有多个茎环结构的长度不一的DNA混合物。
环介导等温扩增的优势与不足
LAMP 的优势:
(1)扩增效率高,能够在1h内有效的扩增1-10个拷贝的目的基因,扩增效率为普通PCR的10 倍-100 倍。
(2)反应时间短,特异性强,不需要特殊的设备。
LAMP 的不足:
(1)对引物的要求特别高。
(2)扩增产物不能用于克隆测序,只能用于判断。
(3)由于其敏感性强,容易形成气溶胶,造成假阳性,影响检测结果。
链置换扩增
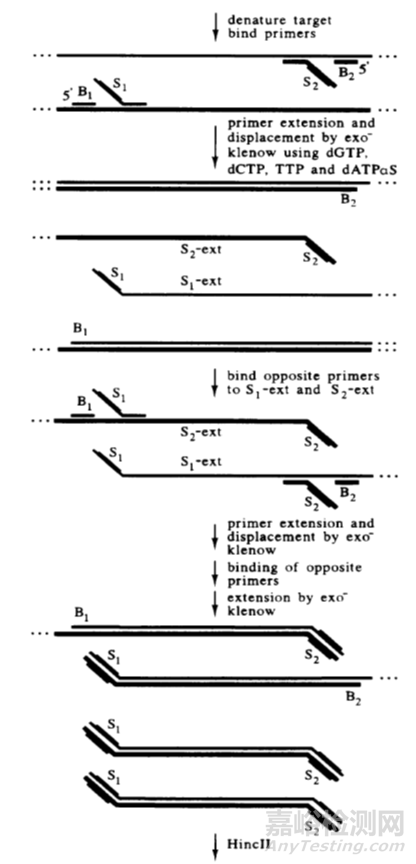
链置换扩增(strand displacement amplification,SDA)是美国学者Walker于1992年首次提出的一种基于酶促反应的DNA体外等温扩增技术。
SDA 的基本系统包括一种限制性核酸内切酶、一种具有链置换活性的DNA聚合酶、两对引物、dNTP以及钙、镁离子和缓冲系统。
链置换扩增其原理是基于在靶DNA两端带有被化学修饰的限制性核酸内切酶识别序列,核酸内切酶在其识别位点将链DNA打开缺口,DNA聚合酶延伸缺口3’端并替换下一条DNA链。
被替换下来的DNA单链可与引物结合并被DNA聚合酶延伸成双链。该过程不断反复进行,使靶序列被高效扩增。
链置换扩增技术的优势与不足
SDA 的优点:
扩增效率高,反应时间短,特异性强,不需要特殊的设备。
SDA 的不足:
产物不均一,在SDA循环中总要产生一些单、双链产物,用电泳法检测时必然会出现拖尾现象。
滚环核酸扩增
滚环核酸扩增(rolling circle amplification, RCA)是通过借鉴病原生物体滚环复制DNA的方式而提出的,指在恒温下以单链环状DNA为模板,在特殊的DNA聚合酶(比如Phi29)的作用下,进行滚环式DNA合成,实现目标基因的扩增。
RCA可分为线性扩增与指数扩增两种形式,线性RCA的效率可达到105倍,而指数RCA的效率可达到109倍。
简单区分,如下图,线性扩增a只用1条引物,指数扩增b则有2条引物。
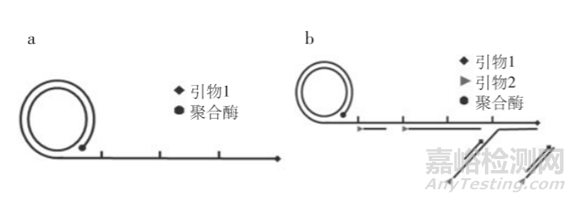
线性RCA 又称为单引物RCA,一条引物结合到环状DNA上,在DNA 聚合酶作用下被延伸,产物为单环长度数千倍的大量重复序列的线状单链。
由于线性RCA的产物始终连接在起始引物上,所以信号易于固定是它的一大优势。
指数RCA,也被称作超分支扩增HRCA(Hyper branched RCA),在指数RCA中,一条引物扩增出RCA产物,第二条引物与RCA产物杂化并延伸,置换已经结合在RCA产物上的下游引物延伸链,反复进行延伸和置换,产生树状的RCA扩增产物。
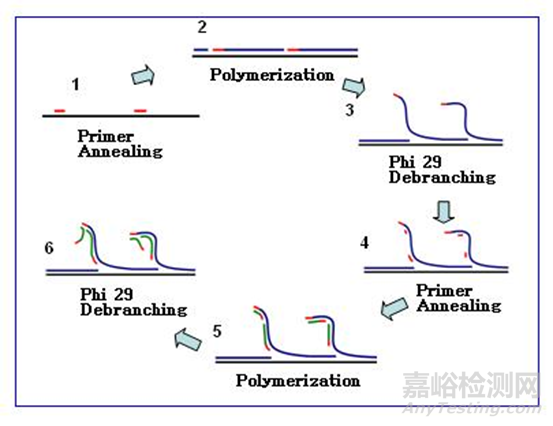
以4BaseBio公司的TruePrime滚环扩增试剂盒为例,使用TthPrimPol引物酶和Phi29DNA聚合酶,实现了无需添加引物即可进行扩增。
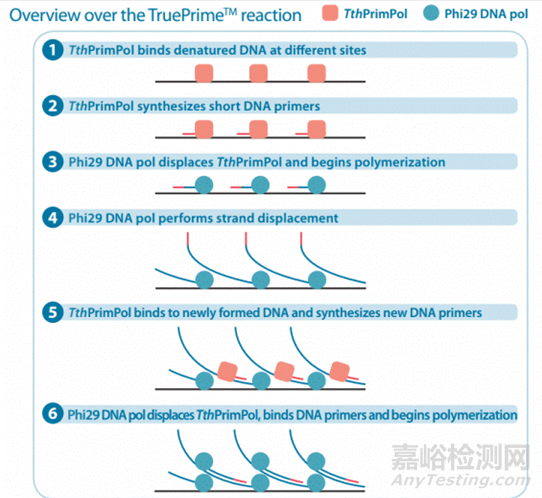
滚环核酸扩增的优势与不足
RCA 的优势:灵敏度高,特异性好,易操作。
RCA 的不足:信号检测时的背景问题。在RCA反应过程中未成环的锁式探针和未结合探针的模板DNA或者RNA 可能产生一些背景信号。
依赖核酸序列的扩增技术
依赖核酸序列的扩增技术(nucleicacid sequence-based amplification, NASBA)是在PCR基础上发展起来的一种新技术,是由1对带有T7启动子序列的引物引导的连续、等温的核酸扩增技术,可以在2h左右将模板RNA扩增约109倍,比常规PCR法高1000倍,不需特殊的仪器。
该技术一出现就被用于疾病的快速诊断,目前有不少公司的RNA检测试剂盒都用此方法。
尽管RNA的扩增也可以使用反转录PCR技术,NASBA相比则有自己的优势:可以在相对恒温的条件下进行,相对传统的PCR技术更为稳定、准确。
反应在41摄氏度下,需要AMV(avian myeloblastosis virus)逆转录酶、RNA酶H、T7 RNA聚合酶和一对引物来完成。
其过程主要包括:
正向引物包含T7启动子互补序列,反应过程中正向引物与RNA链结合,由AMV酶催化形成DNA-RNA双链。
RNA酶H消化杂交双链中的RNA,保留DNA单链。
在反向引物与AMV酶的作用下形成含有T7启动子序列的DNA双链。
在T7 RNA聚合酶的作用下完成转录过程,产生大量目的RNA。
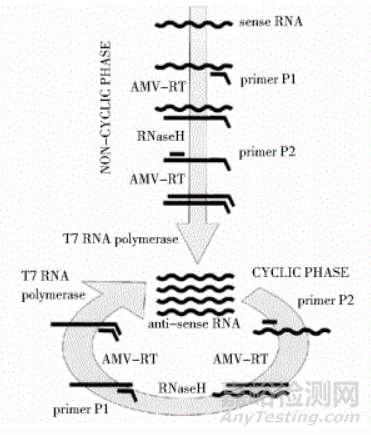
NASBA的优势:
(1)它的引物上带有T7启动子序列,而外来双链DNA无T7启动子序列,不可能被扩增,因此该技术具有较高的特异性和灵敏度。
(2)NASBA将反转录过程直接合并到扩增反应中,缩短了反应时间。
NASBA的劣势:
(1)反应成分比较复杂。
(2)需要3种酶使得反应成本较高。
参考文献:
1.姜苏 , 李一荣,等温扩增技术的原理及应用
2.彭涛,核酸等温扩增技术及其应用
3.赵璐瑶等,基于环介导核酸等温扩增的快检技术研究
4.England biolabs, Loop Mediated Isothermal Amplification.
5.G.TerranceWalker etc. Strand displacement amplification isothermal, in vitro DNA amplification technique.
6.吴晓亮等,DNA的滚环扩增技术研究进展。
7.各公司官网。
03、下篇:测序技术
本篇作为分子诊断技术全解析的最后一篇,介绍一下测序技术。
基因测序技术始于1977年,sanger发明的DNA双脱氧末端终止测序法拉开了序幕。Sanger在1958年和1980年因为胰岛素测序和DNA测序而两度获得诺贝尔化学奖,是第四位两度获得诺贝尔奖以及唯一获得两次诺贝尔化学奖的人。
Sanger测序
Sanger测序采用四个双脱氧核糖核酸(ddNTP)加入到正在合成的链上,由于双脱氧核糖核酸少了一个氧原子,一旦被加入到DNA链上,反应即终止。
通过构建四个反应体系,加入AGCT四种双脱氧核糖核酸,同时调节脱氧核糖核酸(dNTP)和ddNTP的相对浓度,可以使反应扩增得到几百至上千碱基的终止产物。
随后通过凝胶电泳对产物进行分离,分为四个泳道,每个泳道对应一种碱基,然后读取条带显影结果。
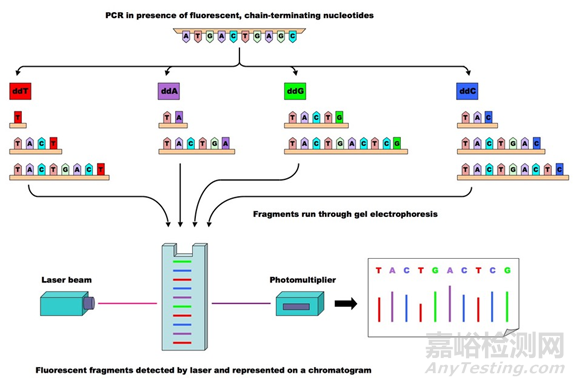
该方法因准确率高,被称为基因检测的金标准,但是耗时长,成本高。
2001年人类基因组计划就是采用sanger测序完成的,从1990年人类基因组计划建立开始,全球的科学家历时11年耗资30亿美元完成。
进入21世纪后,随着物理及化学技术的发展,开始采用相同激发波长但不同发射波长的荧光集团来标记ddNTP,ATGC对应不同的荧光基团产生不同颜色的光被计算机读取,测序的速度和效率大大提高。
二代测序
第二代测序技术也称高通量测序(high-throughput sequencing,HTS)技术,相对于一代测序,它可以实现大规模平行测序,基本原理是将基因组分割成短片段,对短片段测序再进行拼接。
对比第一代测序技术拥有着高通量、低成本等优势,目前相同数据量的检测,其成本约为一代测序技术的0.01%,极大地推动了测序技术在临床检测方面的应用。
2005年454公司基于焦磷酸测序法推出了Genome Sequencer 20 System(GS 20)系统,开启了高通量测序的进程。2007年,罗氏公司收购了454,并推出了一系列性能更优的NGS系统,极大的提升测序通量和准确性。
尽管具有读长优势,但是测序通量和成本始终限制了454平台的推广,同样数据量下成本约是illumina的100倍,因此罗氏在2016年底终止了454NGS测序相关的业务。
2006年Solexa公司推出了Genome Analyzer系统,包括DNA簇、桥式PCR和可逆阻断等技术,这使得GA系统在高通量、低成本、应用范围广等方面具有明显优点。2007年,Illumina公司收购了Solexa并发布二代测序仪。
二代测序经过这些年的发展已经步入成熟期,目前市场上根据测序技术可以可以把二代测序平台分为4类:边合成边测序法(Illumina)、半导体测序法(ThermoFisher)、联合探针锚定聚合测序法(华大智造)和焦磷酸测序法。
Illumina边合成边测序
Illumina测序的流程主要包括样品制备,簇生成,测序,数据分析。
首先是样本制备和建库。用DNA或RNA抽提试剂盒提取核酸,然后用超声波将其随即打断成90-250bp左右的长度或者控制全部DNA在一定长度范围内。
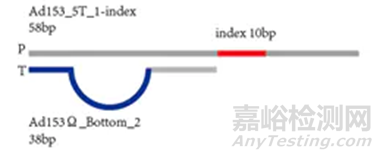
为了后续的扩增和测序,需要在这些DNA片段加入特定的序列。如下图,分别是与流动池引物互补结合的区域(P5、P7)、与Read 1和read 2测序引物结合的区域(Rd1SP,Rd2 SP)以及标签序列区域Index。
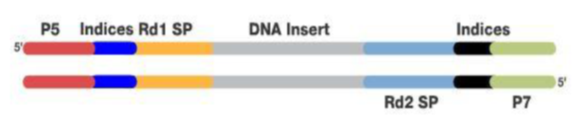
添加完接头序列的DNA合集称为DNA文库,这样就完成了建库,该步骤可以采用商业化的文库制备试剂盒完成。
第二步是成簇Cluster Generation。
成簇是上述DNA片段被扩增的过程,该过程在流动池(Flow cell) 中完成。流动池是一种含有8个通道的厚玻璃片,每条通道中都随即植入了能与文库接头P5或P7互补结合的短DNA片段。
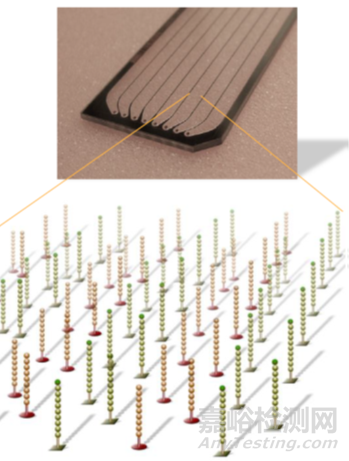
首先引物和流动池的固定DNA片段互补配对,固定在通道表面,然后在DNA聚合酶作用下DNA链进行互补延伸形成DNA双链。通过变性,其中的单链被洗脱,剩下的一条单链会与旁边的固定接头链接,形成单链桥。
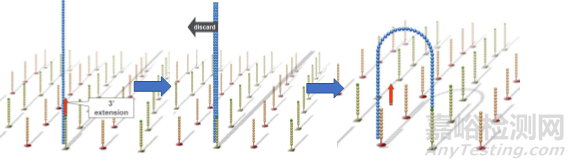
同样的,单链桥在DNA聚合酶作用下延伸配对形成双链桥,通过变性形成2条单链,这两条单链又分别与旁边的固定引物结合,形成2个单链桥。重复这个循环,最终形成数百万的DNA簇。
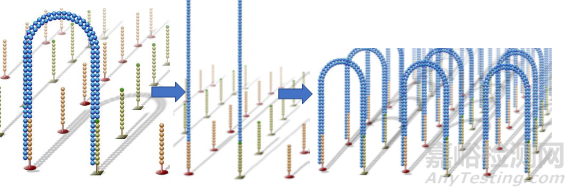
上述过程所有的DNA片段都会被扩增,扩增结束后,反向连会被切断洗脱,只留下正向链,为防止互补结合重新形成单链桥,3‘端被封锁。
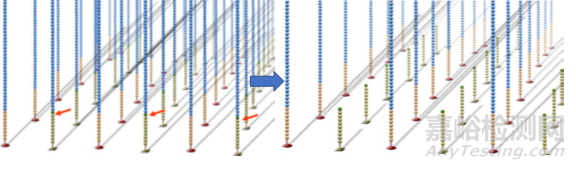
第三步,测序。
首先,在流动池中加入荧光标记的dNTP和酶,由引物起始开始合成子链。由于dNTP存在 3’端叠氮基会阻碍子链延伸,因此每个循环只能测得一个碱基。合成完一个碱基后,洗掉多余的dNTP和酶,使用激光扫描获得荧光信号。
随后加入试剂将叠氮基团与荧光基团切除,然后流动池再通入荧光标记的dNTP和酶,由引物起始开始合成一个碱基。不断重复这个过程,完成第一次读取。
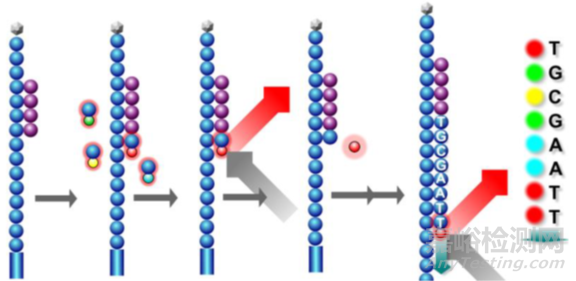
所有的DNA片段的一个碱基会被同时读取,在大规模并行的过程中,机器读取的图像类似下面这样:

同时,加入了不同的index来区分每个样本及正负链。在完成第一次读取后,复制出的链会被洗去,index片段引物被引入并与模板杂交,完成序列读取后被洗去。这样读取到的序列与开始时已知的index比对后就可以给测得的序列贴上标签,方便后续分析。
Paired-end测序已经是现在的主流,要完成双末端测序,首先要将模板链3’去保护,模板折叠,index片段引入,在聚合酶参与下形成双链桥,然后变性,恢复为单链,然后将正向链切除并洗去,留下反向链,与正向链类似,经过多个循环后完成读取。
第四步,数据分析
测序完成后会产生数百万个reads,基于在样品准备时构建的index 分类来自不同样本的序列。对于每个样品来说,具有相似延伸的碱基被聚在一起。正向和反向read配对生成连续序列。这些序列通过与参考基因组匹配后,实现完整序列的构建。
Thermo Fisher半导体测序法
赛默飞的Ion Torrent平台是基于半导体技术的高通量测序仪。该平台使用了一种布满小孔的高密度半导体芯片,一个小孔就是一个测序反应池,孔底部带有感应器。

其测序的核心技术是利用半导体技术进行信息读取,测序过程中每当有碱基结合,便会释放H+,而氢离子会引起电势的改变从而被检测到。通过对氢离子的检测并转化为电信号,最终实现实时碱基判读。
其测序的流程主要包括建库,油包水PCR,测序,数据分析。
首先是建库
与illumina 的 3’端带突出 T 碱基粘性末端的接头有所不同,建库过程中DNA两端加入的是平端接头(P1)和X或A接头,X接头带有index,A接头不带。
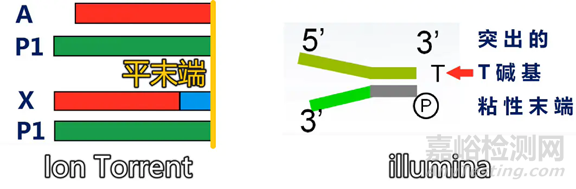
X 接头带Barcode 序列(近末端蓝色序列),而A 接头不带Barcode 序列。
X 接头的好处是可以把一个芯片的测序通量分配给多个文库,测完序之后用Barcode 区分。
A 接头的好处是直接测到样本序列,这样对于充分利用测序的读长无疑是更好的。但是它的缺点是没有Barcode,所以一张芯片只能放一个样本。
AmpliSeq 是Ion Torrent 平台上的建库方案,它的核心是通过多重PCR 的方法,一次从样本中把要测序的多个DNA 片段给扩增出来,然后转化成文库进行测序。
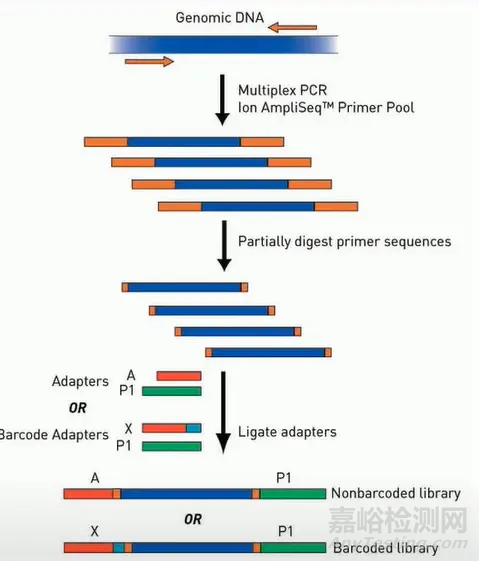
PCR 产物两端20-30bp 碱基都是 PCR引物的序列,如果将其进行测序,则会浪费掉相当一部分测序读长及数据量。因此在这个引物上特别设计了一种化学修饰,这种化学修饰可以被Fupa试剂所消化,而后的测序就可以尽可能多地测到样本序列。
乳液PCR或油包水PCR
Ion Torrent 测序前需要把文库结合到测序微珠上去,并且进行扩增,此种方法称为油包水PCR,也称Emulsion PCR(乳液PCR)。
EP 管中包含油相和水相,其中水相是核心,油相起到分隔作用。水相中包括文库、引物、酶、MasterMix、测序微珠等PCR反应的主要成份。
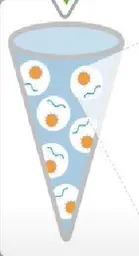
测序微珠的直径约1~2.4微米,每个油包水PCR都含有许多微珠,这些微珠的表面共价连接了许多PCR引物,与P1序列互补。
同时油包水PCR中的游离PCR引物,其序列与A/X接头一致,且5‘端都标记了生物素。
把引物、酶、测序微珠等先在水相中混合,再加入油,混合形成乳浊液,油把水相分隔成一个个的小水滴。PCR反应后,微珠表面就会长出水滴内所含DNA 文库的扩增拷贝。
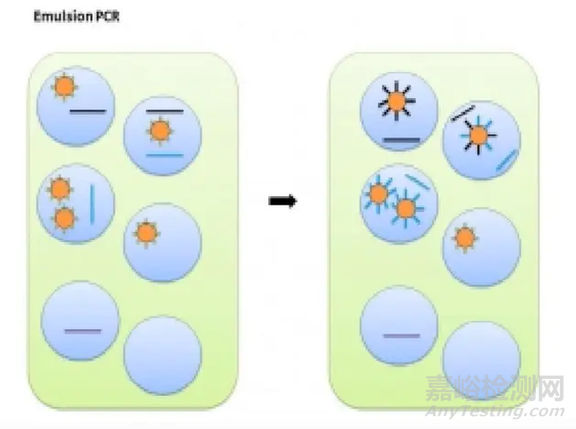
然后加入带有链霉亲和素标记的磁珠与微珠进行混合。发生了PCR 的微珠由于其引物上带有生物素,便会与磁珠结合;没有发生PCR 的微珠由于没有生物素,不会与磁珠结合。接着,用磁铁吸附富集有效微珠,再清洗掉上清液中没有PCR 的微珠,最后用洗脱液把微珠与磁珠分离开来,进行测序。
测序
测序主要发生在一张半导体芯片上,上面做了数以百万、千万计的小孔,每个小孔的既是测序微珠的容器,又同时是一个微型的PH 计。
每个小孔正好可以容纳一个测序微珠,当DNA聚合酶把核苷酸聚合到延伸的DNA链上时,会释放出一个氢离子,反应池中的PH发生改变,位于池下的离子感受器就会感受到信号,把化学信号直接转化为数字信号,从而读出DNA序列。

数据分析
把分别含A、C、G、T 四种 dNTP 的溶液,分别依次地流过芯片的表面。
举例来说,流入的是dCTP 溶液,而模板上正好有一个G 碱基,就发生聚合反应,并产生电压变化,而且会被记录下来。如果流入的溶液与模板上的碱基不匹配,就不会发生聚合反应,也就没有电压变化,也就不会有碱基被记录下来。
如果正好有 2 个一样的碱基相邻,一次就会有2 个碱基被聚合到DNA 链上,电压变化值就会加倍,序列中2 个新的碱基被记录下来。
华大智造联合探针锚定聚合测序法
2013年3月18日,华大基因对CG(Complete Genomics)公司全额收购,开启了华大测序仪之路。历经多年的研发和改进,华大基因相继推出的BGISEQ-500、BGISEQ-50、MGISEQ-200、MGISEQ-2000M、MGISEQ-T7等测序体系。
其测序平台采用的是DNB(DNA Nanoball ,DNA纳米球)测序技术,每个DNA的直径约220-240nm。
主要步骤包括文库制备,cPAS测序,数据分析等。
样本准备和建库
MGI文库构建采用泡状接头及与之对应的扩增产物,有长接头及短接头,单端和双端index。
将文库进行单链分离及环化处理后,以单链环状DNA为模板,在DNA聚合酶作用下使用滚环扩增将单链环状DNA扩增2-3个数量级,此时扩增产物被称为DNB。
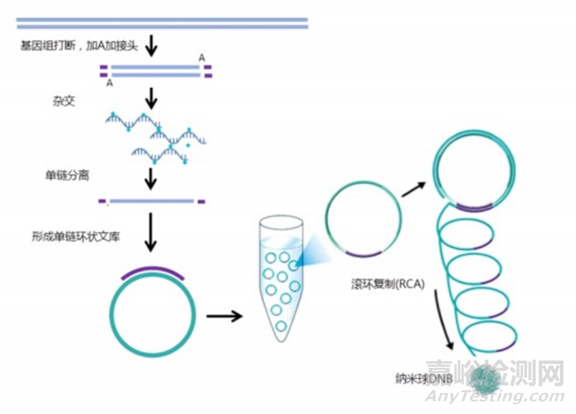
DNB经过DNB装载技术固定在阵列化的硅芯片上形成纳米芯片,由于一个DNB结合到芯片上的小孔后会排斥其他DNB结合,因此每个小孔仅容纳一个DNB,保证了信号点间不会相互干扰。
测序
采用半导体加工工艺,在经过修饰的硅片表面形成结合位点阵列(直径约200nm),实现DNA纳米球的规则排列吸附,阵列位点的间距约700nm,每个位点只固定一个DNB,保证不同纳米球之间的光信号不会互相干扰。
带有荧光探针的Read1引物在DNB上匹配互补,随后系统对光信号采集,得到待测序列。
MDA(multipledisplacement amplification)二链测序,随机引物在多个位点与模板DNA结合,在Phi29DNA聚合酶作用下起始复制,沿着DNA模板合成DNA,同时取代模板互补链;被置换的互补链又变成新的模板进行后续扩增。完成第一链测序后,在该酶作用下形成第二链,通过DNA分子锚,进行第二链cPAS测序。
第三代测序技术
1.PacBio SMRT单分子测序技术
Pacific Biosciences于2004年成立,2010年纳斯达克上市,2011年PacBioRS发布。
以PacBio测序为代表的第三代基因测序技术逐渐应用到多个科研领域。该平台利于单分子实时测序技术,又称作SMRT(Single Molecule Real-Time)测序,基于纳米小孔的单分子读取技术,无需扩增即可快速完成序列读取。
建库
因为测序读长很长,因此可以制备大片段(3-10kb)文库;不同于其他二代测序文库,该文库两端分别连接环状单链,单链两端又与双链正负链连接,得到类似哑铃的结构,称为SMRT Bell。
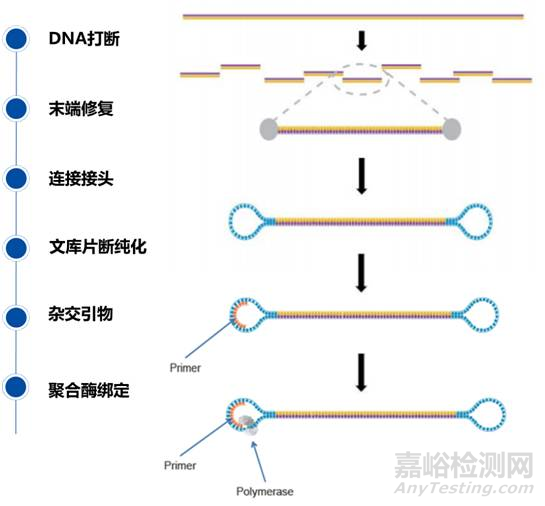
测序
SMRT Cell芯片含有数百万个纳米级的零模波导孔(zero-mode wave guides, ZMWs),ZMW是一个直径只有10~50nm的孔,引物在模板的单链环部位退火后,这个双链部位就可以结合到已固定在ZWM底部的聚合酶上。
每个ZMW都能够包含一个DNA聚合酶及一条DNA样品链进行单分子测序,4种dNTP带有不同的荧光标记。当激光打在ZMW底部时,只能照亮很小的区域,DNA聚合酶就被固定在这个区域。
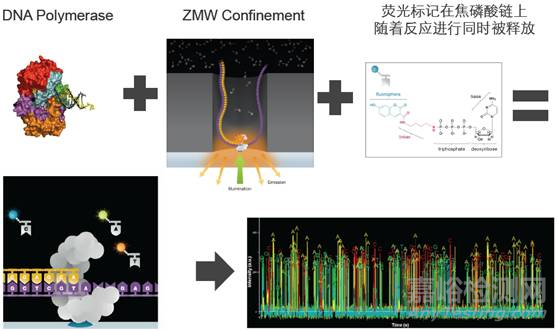
只有在这个区域内,碱基携带的荧光基团被激活从而被检测到。当DNA合成进行时,下一个碱基进行延伸,上一个dNTP上的荧光基团就会被切除,保证了检测的连续性。
不同的碱基会发出不同的光,此时根据光的波长及峰值便可判断碱基类型。
2.纳米孔测序技术
2005年,牛津纳米孔科技有限公司(Oxford Nanopore Technologies)成立,致力于纳米孔测序技术的商业转化。2014年Nano space单分子测序技术发布。
牛津纳米孔公司开发的纳米单分子测序技术与以往的测序技术皆不同,它是基于电信号而不是光信号。
纳米孔测序技术的核心是一种整合了多个跨膜通道蛋白(即纳米孔蛋白)的多聚物膜。通过在膜两侧施加电压从而产生稳定的穿过纳米孔的电流。当有其他物体穿过纳米孔时,会影响电流的大小,从而产生可识别的电信号的变化。
测序时,DNA双链在马达蛋白的牵引下解螺旋为单链DNA,并穿过纳米孔蛋白(也叫Reader蛋白)。由于ATCG四种碱基结构和大小的差异,会使电流产生特征性离子电流变化,通过识别这种电信号的变化,从而达到读取碱基序列的目的。
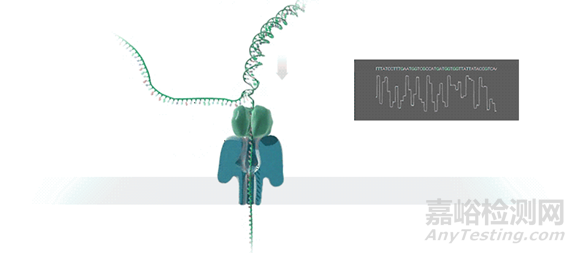
不同型号的纳米孔测序仪原理非常一致,其核心均为纳米孔测序芯片。常规的纳米孔测序芯片整合512个测序通道,每个测序通道包含四个纳米孔,官方数据显示单张芯片不间断测序48h,可产生20-30G的数据量。

二代测序的出现极大地解决了通量问题,在大幅提高测序速度和准确性的同时大大降低了测序成本,但阅读长度相对较短。而以单分子测序为主要特征的三代测序,正朝着单分子、长读长、低成本、小型化的方向发展,实现了测序领域的又一次变革。
参考文献:
1.李金明,《高通量测序技术》
2.各公司官网

来源:Internet


