您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-07-09 18:42
开发一款新药通常要经过DMTA四个阶段,即design(设计)、make(合成)、test(测试)和analyze(分析),其中每一部分都需要大量的时间和资源投入。尤其是在药物合成阶段,在已知目标分子结构的情况下,如何加快目标分子合成路径的设计并减少合成失败的几率,是药物化学家们最关心的问题之一。
计算机辅助合成设计(computer-assisted synthesis planning, 简称CASP)从上世纪60年代就已经出现。早期的CASP软件基于手工编码的反应规则,结合引导启发方法来辅助合成路径的设计,被认为是最早期的人工智能药物合成设计。然而,初代的智能药物合成完全依赖于化学家的专业知识,没有用到基于大量数据的统计学习。直到最近二十年机器学习的兴起,将自动化数据提取和管道式训练引入CASP,使得CASP可以不断扩展来吸收新的反应数据,并集成在自动化流水线中,成为新一代基于人工智能的CASP,即智能药物合成设计。
本文主要介绍智能药物合成设计的主要内容,并以麻省理工大学的ASKCOS作为实例,介绍学术界和业界是如何共同开发智能药物合成设计软件的。
智能药物合成设计的主要内容
智能药物合成设计主要包括三方面的内容:逆合成设计、反应条件推荐和正向反应预测 。
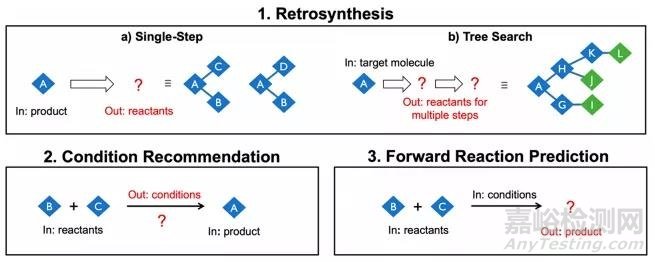
逆合成设计、条件推荐和正向反应预测示意图
逆合成设计(retrosynthetic planning)是智能药物合成设计中最主要的环节。逆合成设计从合成路线的终点即目标分子出发逆流而上,将目标分子断裂成新的小分子(即前体),再在前体上进行新一轮的断裂形成新的前体,如此不断迭代,直到所有的前体化合物都是市面上有售的化学原料为止。这样的迭代过程对药物化学家是一个难题,因为人脑无法同时对大量的分子进行评估,也无法同时处理多条假设的合成路线。相反,迭代问题是计算机擅长的领域。计算机可以通过各种树搜索算法的使用,将单步逆合成扩展到全路线设计,每一步可以产生数千个前体。另一方面,由于合成路线的终点往往由分子是否能被买到决定,目标分子合成能力的评估并不是一个基于分子结构的平滑方程,而逆合成软件可以通过神经网络模型来模拟这样的非线性方程。
有了合成路线之后,还需要考虑实验室中进行每一步合成的可行性。人们希望智能药物合成软件也能够推荐反应条件,减少经验筛选耗费的时间。然而,通过人工智能推荐反应条件的难度在于,一个完整的反应条件包括反应物的数量、质量或浓度、反应时间、试剂与催化剂的加入顺序等很多的细节,然而大部分的反应数据不能完整地记录这些细节。此外,反应条件的推荐与反应的目的和评估条件也有关联。有些反应人们追求反应物的转化率,而另一些反应人们希望尽量降低副产物的生成。因此,反应条件的推荐往往需要结合反应优化这一更为成熟的领域。反应优化以反应条件做变量来构建反应性能的模型,而机器学习可以通过各种搜索算法来加快模型的优化并提供不确定性的评估。
正向反应预测通过预测反应产物来确保合成路线设计的可操作性。利用机器学习的反应预测在近几年得到了较快的发展,主要有基于反应规则与模板的预测、图神经网络预测原子和化学键从反应物到产物的变化、基于自然语言处理的SMILES产物预测三大方法。正向反应预测的主要作用是预测副产物的生成和反应的选择性。将前向反应预测和逆向合成设计结合,可以用前向反应预测来评估逆合成设计。逆合成设计中每一个单步骤合成都有可能存在可替换的起始材料集合,前向反应预测可以对这些集合进行排序,从而选出最佳的方案。
智能药物合成设计的实例分析:ASKCOS
美国麻省理工大学与十余家大型制药与生物科技公司合作,成立了“药物发现与合成的机器学习联盟”(Machine Learning for Pharmaceutical Discover and Synthesis Consortium, 简称MLPDS),旨在促进药物自动化发现与合成软件的开发。该联盟的实力处于全球领先水平,其中麻省理工大学化学工程系的Coley和Jensen团队是最早开始研究人工智能应用于化学反应预测及逆合成分析的团队之一,而联盟中的企业成员则包括阿利斯康、巴斯夫、拜耳、葛兰素史克、礼来、默克、诺华、辉瑞、药明康德等巨头。联盟开发的开源智能药物合成设计软件ASKCOS被应用在成员公司的DMTA工作流程中,而成员公司通过对ASKCOS的功能提出反馈来促进ASKCOS的进一步开发。
ASKCOS提供了独立的图形用户界面,使得化学家们可以轻松地与ASKCOS建议的路线和预测进行交互。不同公司和不同部门对ASKCOS使用的侧重点不一。有些化学家通过同时使用合成设计工具和传统的数据库来查找已知反应,以便快速地产生新的想法,而计算化学家和化学工程师则更多地关注分子设计和工艺开发。这样的好处在于,人们可以从不同角度对ASKCOS的性能进行评估,并且对模型的建议进行验证。因此,对使用人员进行基础培训非常重要,包括介绍软件背后的理论以及如何使用软件中不同的模块,并传达给使用人员这样一个信息:ASKCOS的目标是通过已知反应数据进行合成路线的预测,而不是作为一个搜索软件用来查找已知的合成路径。反之,如果没有这些必要的培训,化学家很有可能带有偏见地去尝试ASKCOS,比如输入一个自己喜欢的化合物,却没有得到已知的合成路径,就很可能劝阻大家不要再使用这些软件。因此,开发人员与药物化学专家的交流是ASKCOS能快速进步的基本条件。
ASKCOS最重要的功能是多步合成路线设计,而使用结果表明,合成路径设计能否成功的主要因素在于可用化合物数据库的覆盖范围。换句话说,公司能购买到的化合物越多,成功获得合成路径的可能性就越大。葛兰史素克公司发现,通过ASKCOS设计69个目标分子的合成,如果采用公开的化合物数据库(含138k化合物),54%的分子发现了可用的合成路径。而使用公司内部的扩展数据库(含8M化合物),则ASKCOS可以为67%的分子的规划可用合成路径。这种现象是由智能算法中搜索成功的条件决定的,如前文所述,通常搜索的终止条件是化合物的商业可用性,即该材料是否能被买到。此外,此现象给了人们一种启发,因为初始的CASP系统往往是根据公用的数据集, 如Reaxys和USPTO。如果公司可以基于自己内部的数据库对ASKCOS进行进一步的训练,则可以对多步合成路线的设计产生积极的影响,并且公司内部的数据库往往比公开的数据库更为重要。礼来公司的数据表明,当采用公司内部的模板集(含13297个模板)训练其内部规划平台ChemoPrint,成功提供路线的几率为40.1%,而加入了公开的专利数据(50275个模板)后,其成功率仅提高了5.8%。
ASKCOS的交互性在自动设计出现问题时起到了关键作用。ASKCOS可以在单步合成预测中实现交互式路径规划。一旦自动合成路径设计失败,专家可以和软件进行交互式探索。ASKCOS可以将类似的建议根据K-means聚类算法进行归类,然后邀请化学家介入,将已归类的多种路径可视化显示出来供化学家参考。
除了多步合成路径设计,ASKCOS也具有正向反应预测和反应条件建议的功能。正向反应预测的目的是验证全路径设计提供的路径,主要用来识别潜在的副产物和杂质。与逆合成设计类似,公司也可以通过内部的反应数据对模型进行进一步训练,以提高特定化学反应的准确性。反应条件的建议功能由于受到有限的训练数据的限制,被采用的机会较低。化学家们通常通过这个模型来确认自己提出的条件,或者加以简单的评估和建议来向开发者提供反馈。
ASKCOS在逆合成分析中的案例之一是Branebrutinib的逆合成设计。虽然该化合物的合成路线已在2016年被文献报道,但当时ASKCOS的训练数据中并没有加入这一路线。ASKCOS采用与文献报道类似的起始材料,提出Boc保护烷基酰胺中间体3的N-H键,并建议通过3和4的C-N偶联来合成1。文献报道中作者提出了他们尝试过C-N偶联,但发现甲酰胺的存在阻止了偶联的进行,因此使用了一个腈类的类似物质来替代4。虽然ASKCOS没能像化学家一样捕捉到这一细节,但它提出的想法和化学家最初的尝试不谋而合,并且它提出的分子断裂也是合理的。
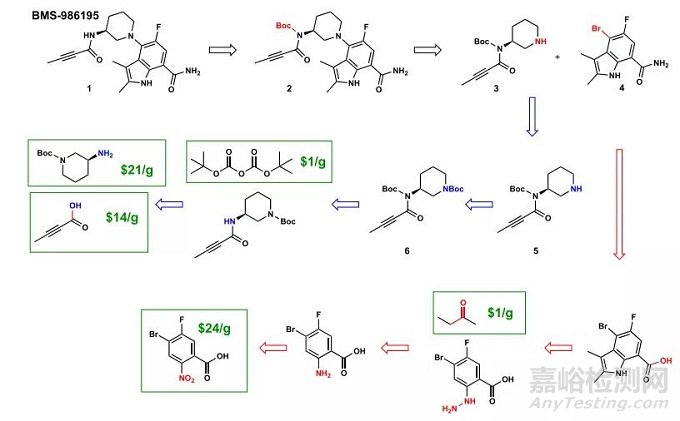
ASKCOS对branebrutinib的逆合成分析
智能药物合成设计的难点
ASKCOS的逆合成分析案例与文献报道的差异揭示了智能药物合成设计软件开发中的一大难点:数据偏见。基于文献的化学反应数据库通常只包括最终成功的反应路径和起始材料,而在设计过程中因为各种原因而被替换的路径和材料则很少会收录到数据库中,但这些信息对化学家和智能软件设计合成路线都非常有用。同时,化学反应数据库通常只包含高收益的正面数据(成功的反应),却很少记录负面数据(失败的反应),导致模型很难去预测一个反应失败的可能性。此外由于分析化学物质的高成本,不是所有的研究都会把所有副产物逐一分析,而副产物的缺失将导致化学反应数据本身的不完整。同时,有很多已经获得的但没有公开的实验数据,尤其是高通量实验数据没有被公开和分享,或者数据的记录格式不适合用计算机读取,使数据无法及时收录到公共数据库中。
智能药物合成设计的另一大难题是评估标准的统一。在模型准确率的评估中,Top-k准确率是常用的指标之一。在Top-k准确率中,标准答案在模型推荐的得分最高的k个答案中即判定为准确。然而,k值大小的选择存在着矛盾性。Top-1准确率虽然可以简化模型的评估,但它与逆合成设计的多样性相违背。在模型训练中,模型推荐了多个正确路线中的一个,但却因为和文献报道的 “标准答案” 不一致导致模型被惩罚,无疑会增大模型训练的难度。相反,如果使用太大的k,如Top-10准确率,又会夸大模型的准确性。模型预测的多样性是另一项重要的评估标准。在不同的应用场景,人们需要不同的推荐方案。在时间紧迫的情况下,一个创新性很高但不够实用的合成路线可能还不如另一个平平无奇但可行性高的路线。但是有些时候人们可能不想设计出已被文献报道过的合成路径,希望看到不一样的推荐。不同的角色对推荐方案的要求也不同。药物化学家可能更希望看到具有共同中间体的路线,从而获得多样的目标分子,但对于每个分子来说该路线都不是最佳的路线。相反对于过程化学家来说,他们更希望看到高度趋同的路线,因为他们需要在合成路线之外的地方花更多的精力(如工艺优化)。
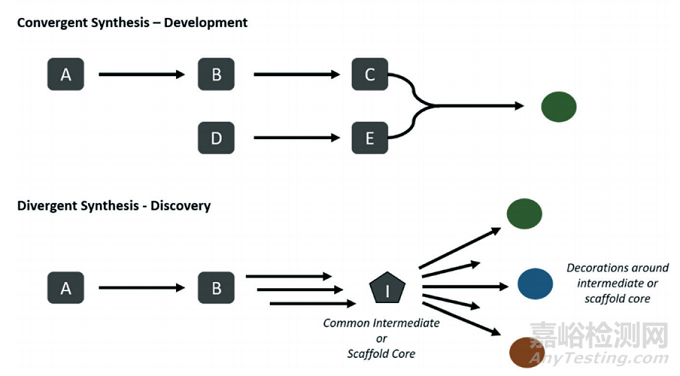
药物开发与药物发现所期望的不同风格的合成路线设计
条件推荐系统发展的相对滞后也会对智能药物合成设计造成很大的影响。条件推荐系统的数据匮乏,主要原因之一是催化反应的数据有限。相对于起始材料广阔的化学空间,催化剂的化学空间要小很多,另外,而在药物合成路线的设计中,不必要地使用催化剂是不被允许的,因为催化剂和配体的合成和表征将额外消耗大量的时间。因此开发基于小数据集、针对催化剂的机器学习模型也非常有必要。
与其他领域的机器学习模型一样,智能药物合成设计软件的可解释性也有待加强。如果软件可以对自己的预测进行解释,那无疑可以增加人们对它的信任,可以吸引更多的人来使用这些软件。同时,模型也需要增强对不确定性的估计。必要的不确定性分析可以帮助化学家意识到什么时候可以完全信任软件的预测,而什么时候应该人工介入参与设计和评估。
展 望
所有的智能药物合成设计,其最终目标都不是替代化学家,而是减轻化学家在合成设计中的认知负担,将化学家从重复性的、不需要太多智力的劳动中解放出来,使化学家们可以将精力集中在一些稀有化合物和复杂反应的研究中。即使每个化学家只将10%的工作量分给计算机去做,也将显著地节省总的时间成本。

来源:Internet


