您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-11-01 23:06
基于片段的药物设计(fragment-baseddrug design, FBDD)是一种将随机筛选和基于结构的药物设计有机结合的药物发现新方法。基于片段的药物设计的方法首先筛选得到低分子量和低亲和力的片段,然后基于靶点结构信息进行优化或者连接,得到与靶点亲和力更高且成药性更好的新分子。在此过程中添加基团是重要的片段组装和优化手段。近十年来,随着片段检测、筛选和组装技术的不断成熟和完善,基于片段的药物设计方法逐步从理论走向实践,并且取得飞速发展。
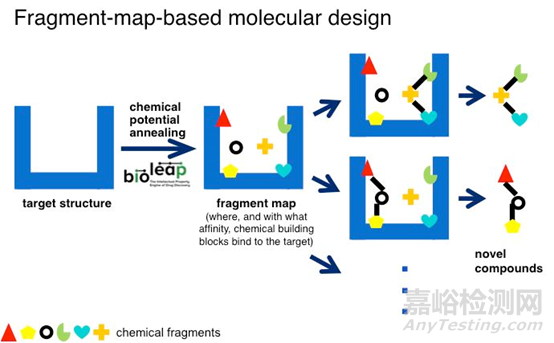
图1.基于片段药物设计的基本原理
1981年,Jencks等首次提出了基于片段药物设计的基本设想,研究认为,药物分子结构中的每个片段都在药物靶标结合过程中发挥着自身作用,因此将不同的结构片段进行组合或者延伸,可以得到高活性的新分子。理论研究证实,将作用于药靶结合口袋不同区域的片段连接成新分子后,会引起结合自由能跳跃式地下降,从而导致亲和力有大幅度地提高。随后,Nakamura等于1985年用该方法设计HMG-CoA还原酶抑制剂,为该理论提供了实验基础,但是在当时而言,与靶蛋白结合片段的识别,片段连接和结构优化仍然是巨大的挑战。1996年,基于片段的药物设计真正取得了突破,Shuker等首次提出了核磁共振构效关系研究方法(SAR by NMOL/LR),该研究小组首先用核磁共振技术检测得到了FK506蛋白的两个低亲和力片段,然后通过片段连接和优化发现了高户型FK506蛋白质抑制剂。自此以后,基于片段的药物设计方法开始引起大制药公司和学术界的广泛关注,并且得到了飞速发展。
一般来说,基于片段分子的设计研究可以分为3个阶段:片段筛选、片段与药靶复合物结构确证、基于片段构建新分子。首先,采用灵敏的检测技术筛选片段库,发现能与药靶结合的片段。片段与药靶的结合力一般比较弱,通常为毫摩尔级别。其次,需要确定片段与药靶是如何相互作用的。最后,根据片段与药靶相互作用的结构信息来指导片段优化片段,或者将作用与药靶活性口袋片段进行连接,构建新分子。设计得到新分子后,通过化学合成手段得到实体化合物,进行生物活性评价,探讨构效关系,发现高活性的新化合物实体。
与高通量筛选等传统的新药筛选等方法相比,FDD的有点主要体现在如下三个方面:
(1)可以探索更为广阔的化学空间。类药(drug-like)小分子的化学空间约为1060,而目前及时最全的化合物库也仅有105~106个分子,仅占类药分子空间极小部分,决定了高通量筛选发现活性化合物的高绿不会很高。儿对于片段分子,符合三原则的片段数目估计在107个左右,现有片段库含有约103~104个分子,占总片段库化学空间的比例药大得多,因此从先天上决定了基于片段的药物设计方法发现活性化合物的概率要高于高通量筛选。
(2)发现活性化合物的命中率高。片段分子具有体积小和复杂程度低的特征,理论上更容易与药靶结合,加上片段筛选技术的灵敏度高,因此具有较好的命中率。
(3)发现新药的可行性高。一般来说,将先导化合物优化至候选药物的过程会伴随着分子量和疏水性的升高,片段分子的分子量范围在120~250。在片段优化至候选药物的优化过程中,分子量和生物活性呈线性增长关系,结构优化空间较大,可行性更强。
01、活性片段分子的发现
(1)片段库的建立
一个高质量的片段库是进行基于片段药物设计的前提条件。构建片段库需要考虑三个因素:库容量、化学结构多样性和类药性。鉴于片段分子的分子量比较小,复杂程度低,片段的容量一般在1000~10000个之间。片段库所含片段应具有较好的化学多样性。这样可以搜寻更大范围的化学空间。类药性方面,片段应该符合“三原则”:片段分子量应该小于300,氢键供体或者受体的数目小于或者等于3个,脂水分配系数(log P)小于等于3。
(2)片段库的筛选
片段库构建完成后,最关键的步骤就是筛选和识别与靶蛋白结合的片段,目前常用的识别技术有:生物化学检测、表面等离子共振技术(SPR)、核磁共振技术(NMOL/LR)、质谱和X射线衍射。
(3)结构信息的确定
确定片段结合信息对知道结构优化至关重要,在片段检测方法中X射线、核磁共振和质谱可以直接或者间接地进行结构信息的测定
02、活性片段分子的优化
从新药发现的角度而言,发现活性片段仅仅是研究的第一步,将活性片段转化为先导化合物甚至是候选药物才是基于片段药物设计的最终目的。活性片段虽然活性较弱,但是它为先导化合物的发现提供了一个高质量的起始结构。片段的优化研究通常在活性片段和其与靶蛋白作用信息基础上进行,可合成性也是重点考虑的问题。从片段到先导化合物的设计方法主要有:片段生长法(fragment growth)、片段连接法(fragment linking)、片段自组装法(fragment self-assembly)。
(1)片段生长法
片段生长又称为片段演化或者片段加工,其原理如下图所。活性片段a分子量小,结合力弱,基于基团添加理念,在片段a的基础上接入合适的基团或者小分子片段,使得到的新分子能同时结合在片段a毗邻的活性口袋,从而提高分子活性,改善理化性质。
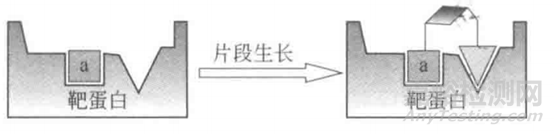
图2.片段生长原理
过氧化酶体增值物激活受体(PPAR)是治疗II型糖尿病的重要靶点,它有三种亚型:PPARa、PPARγ、PPARδ。Plexxikon公司的科研小组利用晶体筛选方法筛选小分子片段库(分子量在150~350之间),初步筛选得到170个活性片段。其中分子片段4-48的结合方式与其它已知配体不同,其吲哚环上1位N原子指向毗邻的一个重要结合口袋。在N原子上连接一个侧链,使其结合到毗邻的结合口袋得到苯磺胺类化合物4-49,如下图所示。根据后者的结构和作用方式,研究人员合成了20个氨苯磺胺侧链的类似物,经过生化实验和细胞活实验的检测,最终优化得到候选药物indelitazar(4-50),indelitazar是PPARα的完全激动剂和PPARγ的部分激动剂,降低了因完全激动PPARγ造成的副作用,目前,该化合物已经处于临床试验阶段。
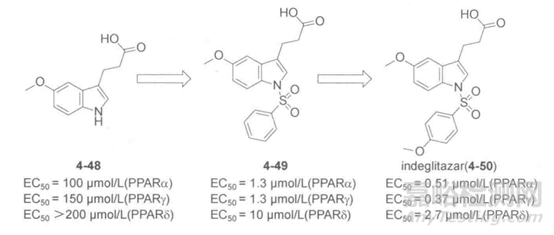
图3.PPAR激动剂结构和活性
周期素依赖性蛋白激酶2(CDK2)是细胞周期中重要的调控元件,也是癌症治疗的重要靶标,Astex公司采用高通量X射线晶体学筛选能在CDK2铰链区(Glu81和Leu83)至少形成一个氢键的分子片段,发现片段4-51能够满足要求,采用基于结构的药物设计侧率优化片段得到化合物4-52,进一步优化得到化合物4-53,如下图所示。晶体结构表明,该化合物能够和CDK2的铰链区的Glu81和Leu83形成氢键,与Asp145形成水分子介导的氢键作用,活性得到极大地提高。研究人员从该分子选择性、细胞活性和药代动力学进行优化,得到了化合物4-54,该化合物已经进入临床试验。
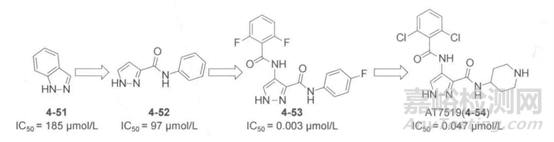
图4.CDK2抑制剂结构与活性
凝血因子Xa是治疗凝血障碍的重要靶点。Protherics的研究人员X射线晶体学方法筛选片段库得到活性片段4-55,该片段在凝血因子Xa的S1口袋,与Asp189形成氢键。以此为模板,研究人员基于凝血因子Xa活性腔结构设计了三类药效团模型,以此模型对ACD库进行虚拟筛选,通过分子对接优选分子,生化试验测试活性,最终发现化合物4-56具有良好的抗血栓活性,如下图所示。但是该化合物口服生物利用度低,推测可能是苯甲脒基团对吸收和分布带来不良影响。研究人员将其用吲哚替换,得到化合物4-57(LY-517717)。LY517717的Ki值为5nmol/L,并且具有良好的药代动力学性质,目前已经进入临床试验。
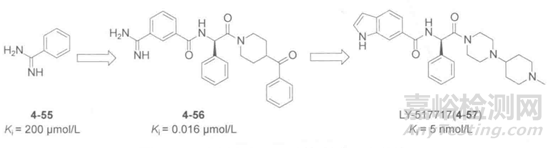
图5.Xa抑制剂结构及其活性
(2)片段连接与融合
片段连接的原理下图所示,片段a和b分别作用在靶蛋白的不同活性口袋,且活性口袋相邻,将俩个片段连接得到亲和力更强的新分子,如果两个活性片段结合的位点有部分重合,这样可以将重合部分以合适的方式合并。一般来说,两个毫摩尔级别片段能够融合成具有微摩尔级别的分子,这种现象叫加和效应(additivity effect)。下面结合具体的实例来介绍片段连接和融合方法的运用。
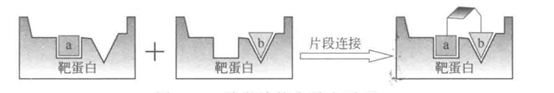
图6.片段连接和融合原理
肿瘤发生于Bcl-2和Bcl-XL的过表达相关,研制Bcl-X1的高效抑制剂对癌症的治疗有重要的意义,Abbott公司用HTS筛选Bcl-XL抑制剂未获得成功。随后,他们采用NMOL/LR技术筛选片段库得到活性片段4-58和4-59;它们分别作用在相邻的两个活性位点,根据它们与Bcl-XL结合的晶体结构复合体,研究人员移除片段4-59的羧基,用磺酰基连接片段4-58和4-59,经结构优化得纳摩尔级别的化合物4-60,如下图所示。后者经过分子选择性和药代动力学性质优化得到了ABT-263(4-61),进入了临床试验研究。
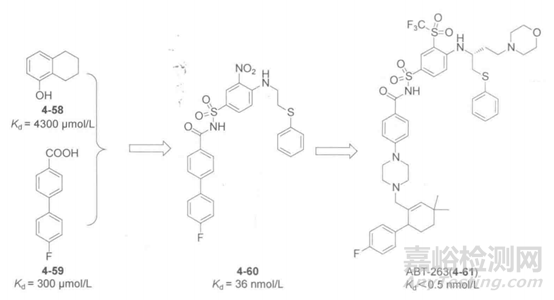
图7.Bcl-XL抑制剂活性与结构
基质金属蛋白酶(MMPs)也是抗肿瘤药物靶点,Abbott公司研究小组用NMOL/LR法筛选片段库,发现片段4-62和4-63分别结合与Zn离子口袋和S1口袋,解离常数分别为17 mmol/L和0.02 mmol/L。根据这两个片段在MMP活性部位的结构和位置,发现他们可以用两个亚甲基连接得到新分子4-64,如下图所示,后者活性得到显著提高,其Kd值达到了15 nmol/L,对其进行进一步修饰得到了新的对MMP-2和MMP-9具有良好活性的4-64,该化合物也进入临床试验阶段。
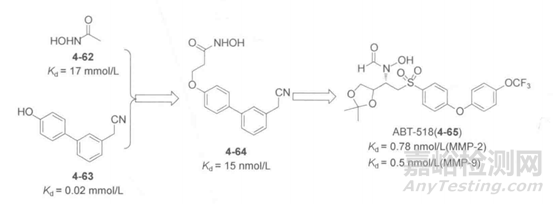
图8.基质金属蛋白酶抑制剂及其活性
丝氨酸蛋白酶尿激酶是一个重要的抗肿瘤作用靶点,Abbott公司利用X射线晶体方法学筛选与尿激酶相互作用的小分子片段,得到了活性片段8-羟基-2-氨基喹啉(4-66),然而,该片段经过结构优化未能如期得到理想化合物,研究人员另辟蹊径,发现化合物4-67是已经发现的尿激酶抑制剂,与化合物4-66作用在尿激酶的相同的活性口袋,但是口服利用度极低,研究人员将化合物4-66余4-67进行融合得到了4-68,如下图所示,其活性达到了0.37 μmol/L,口服生物利用度也提高到了38%。
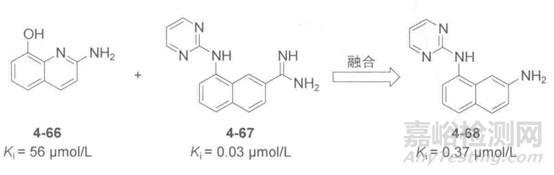
图9.尿激酶抑制剂结构及其活性

来源:化学经纬


