您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-11-29 19:44

注:新的《医疗器械监督管理条例((第739号令))已于2021年5月1日实施》
图1 产品技术要求的地位
2014年6月1日颁布实施的《医疗器械监督管理条例》(国务院令第650号,以下简称《条例》),将产品技术要求取代了产品标准,明确了产品技术要求的法律地位。见图1。
早期企业注册医疗器械时,递交的产品注册资料是产品标准,当产品获批后该标准就转化为产品注册标准,很多企业习惯了用产品注册标准代替成品检验标准,在产品注册标准中规定了其中若干个项目作为出厂检验项,然后全检项目为型式检验项目,这样无形中就将出厂检验标准降低了,显然,这种做法是不合理的,其无法可靠保证售出产品安全、有效。
《规范》
2015年3月1日实施的医疗器械生产质量管理规范的公告(2014年第64号以下简称《规范》)中第五十八条 企业应当根据强制性标准以及经注册或备案的产品技术要求制定产品的检验规程,并出具相应的检验报告或证书。
规范明确通过产品检验规程涵盖强制性标准以及经注册或者备案的产品技术要求的性能指标;要求检验记录能够证实产品符合要求;并根据检验规程及检验结果出具相应的检验报告或证书。
2016年3月1日《总局办公厅关于医疗器械产品技术要求有关问题的通知》中有关事项通知中的(五)明确:产品技术要求主要包括医疗器械成品的性能指标和检验方法,其中哪些项目需要出厂检验,不在产品技术要求中规定。企业应当根据产品技术要求、产品特性、生产工艺、生产过程、质量管理体系等确定生产过程中各个环节的检验项目,最终以产品检验规程的形式予以细化和固化,用以指导企业的出厂检验和放行工作,确保出厂的产品质量符合强制性标准以及经注册或者备案的产品技术要求。
2017年1月4日,《医疗器械生产企业质量控制及成品放行指南(2016第173号)》,进一步推动规范及其附录的有效实施,指导医疗器械企业按照产品技术要求做好质量管理工作,强化采购、生产、检验过程中的治疗控制,帮助企业科学、合理的制定出厂检验规则,严格医疗器械成品放行,从而提升医疗器械整体质量安全水平。本指南就产品实现全过程,特别是采购和生产过程的质量控制、成品放行具体内容和要求进行适度细化,提高了《规范》要求的可操作性。见表1。
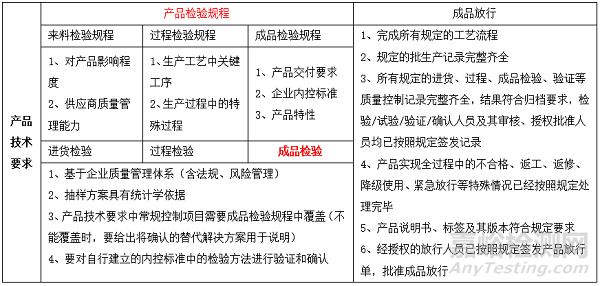
表1 产品技术要求与产品检验规程转换关系
为制定符合公司产品技术要求的检验规程,明确成品检验项目。针对产品技术要求的性能指标,结合迈瑞公司质量管理的现状,依据当前技术水平或行业通行做法,将成品检验的检验项目分为非常规控制检验项目和常规控制检验项目。
制定如下规则:
a) 完整性:
应当对每一份产品技术要求进行设计转换;
应当对《产品技术要求》中所有性能指标进行转换说明。
b) 合理性:
应依据当前行业技术水平或通行做法、产品的性能和功能等指标受生产过程影响程度;
检验规程原则上应当覆盖已注册或者备案的《产品技术要求》中需要常规控制的检验项目和检验方法。不能覆盖的,应当予以说明;
检验方法与产品技术要求中的检验方法不一致的,应予以说明。
根据以上原则,建立成品检验控制方案和产品检验规程模板见图2、表3、表4。
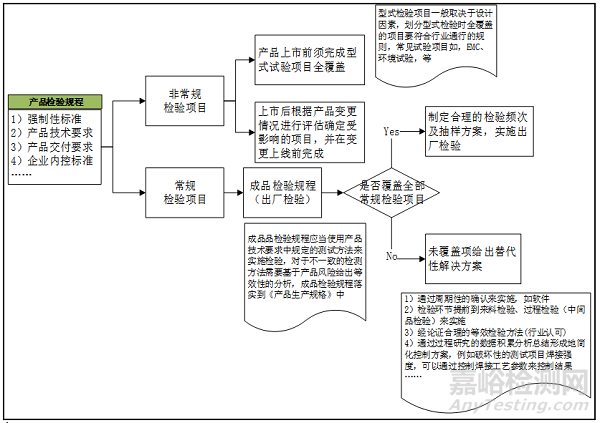
图2 成品检验控制方案
|
非常规检验项 |
|
1. 检验项目不适宜或无法在大批量连续生产的情况下实施的,可包括以下场景:
Ø 检验需要重新对机器外壳等进行拆除才能进行的;例如,某些安全类测试; Ø 检验有可能对产品造成潜在不良影响或损害的;例如,环境试验,静电测试等; Ø 检验需要特殊设备或场地才能实施的;例如,环境试验,EMC试验等; Ø 检验需要有特殊资质的人员才能实施的;例如,针对强制性标准的覆盖检测; Ø 其他需要添加的场景
2. 不受生产过程影响的软件功能(产品技术要求中体现为软件功能,检验方法为只对软件功能进行检查,不涉及产品性能)。 |
表3 成品检验中非常规性说明
|
|
||||||
|
产品技术要求 内容 |
是否 常规控制项 |
出厂检验控制 |
转换 说明 |
生产 规格 |
||
|
控制节点 |
全检 |
抽检 |
||||
|
1 |
|
|
|
|
|
|
|
1.1 |
|
|
|
|
|
|
|
1.2 |
|
|
|
|
|
|
|
…… |
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
2.1 |
|
|||||
表4 产品检验规程模板

来源:德大器械产业管家


