您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-12-04 20:43
01、医疗器械新法规MDR
新的医疗器械法规 (MDR, EU 2017/745) 引入了一些新的、重大变更的流程,如果制造商已经或打算在欧盟销售医疗器械,则必须将以下流程纳入制造商的质量管理体系 (QMS)。
1.质量手册内容引用的欧盟标准和MDR法规;
2.确定法规质量负责人的资格及责任权限;
3.建立售后市场监督管理制度(PMS);
4.设计开发过程的文件变化
5.修订CE技术文件相关的程序文件;警告系统、不良事件报告程序和申报EUDAMED程序;修订临床评估程序文件;修订产品的分类依据以及CE认证的模式;修订上市后的临床跟踪程序文件(PMCF);建立欧盟UDI程序文件并导入UDI实施;修订售后、事故、投诉及欧代相关的程序文件。
02、ISO 13485:2016 与MDR要求之间的对应关系图

符合MDR的设计开发过程的要求,下图给出了在设计开发阶段必须要写的其他设计记录
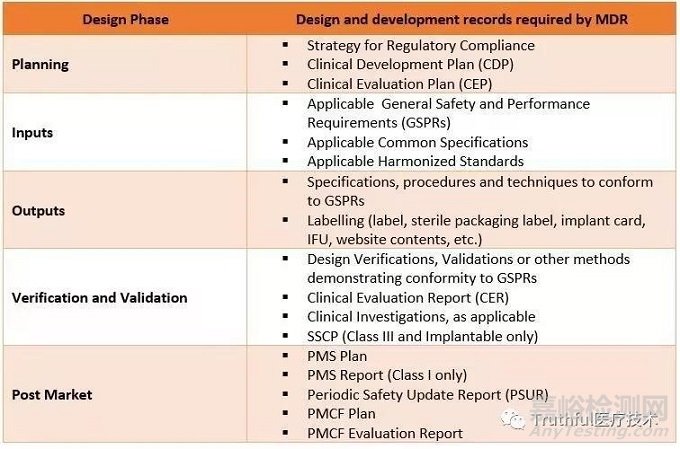
03、CE技术文档内容
1.签名及受控编号等内容;
2.DOC声明文件,包括UDI代码、新指令、分类途径、认证模式、欧代等;
3.医疗器械的描述、新法规下性能指标的改进、包括设计变更、附件、标签及说明书等内容;
4.MDD/MDR法规下,产品相关性能参数的对比;
5.制造商信息、工艺、设计及制造信息的变更;
6.新法规检查表、通用安全及性能要求CS等;
7.产品验证与确认,包括工艺变更、过程检验、出厂检验等,以及可能包含软件部分;
8.测试报告(包括可能的电气、生物学、性能等)
9.风险分析等(很快会实施ISO14971:2019版本),变化较大;
10.MDR下MED2.7.1 4.0临床第四版,临床性能、临床数据、性能评估及统计分析等;
11.上市后监督(PMS)及警戒系统等具体文件;
12.稳定性实验、性能验证(包括理化等)、运输试验;
13.有源产品软件的验证与可用性,带无线的产品还需要考虑无线共存,数据包丢失等;
14.定期安全性更新报告PSUR,引用的国家标准法规等;
15.欧孟授权代表:欧代需要具备相应资质,欧代与制造商出口商同责,购买商业保险、重新签署MDR的欧代协议、明确欧代的责任和义务等
上述CE ISO更新的内容均为重要内容,MDR不是简单的更新一些分类或认证模式,而是从产品的工艺上考虑相关的变化。

来源:Internet


