您当前的位置:检测资讯 > 行业研究
嘉峪检测网 2021-12-21 22:38
摘要 目的:系统了解美国孤儿药批准情况,分析资格认定和审批趋势。方法:以美国食品药品监督管理局(FDA)数据库Drugs@ FDA和孤儿药资格认定和批准在线数据库为主要数据来源,系统收集美国FDA自1983至2020年认定和批准的孤儿药信息,从审批数量、审批时间、治疗领域等方面进行统计分析。结果:1983-2020年FDA共认定孤儿药资格5757个,批准孤儿药适应症943个。从孤儿药认定身份到获批上市平均所需的时间为5.14年。批准的孤儿药适应症中抗肿瘤和免疫机能调节相关适应症最多,为408个,占比为43.27%。结论:自1983年《孤儿药法案》实施以来,FDA授予的孤儿药资格数量和批准的孤儿药适应症数量呈现明显的增长趋势。
孤儿药,又名罕见病药,是指用于治疗、预防、诊断罕见病的药品。世界各国对罕见病的认定标准存在一定的差异。美国将罕见病定义为每年患病人数少于20万人的疾病 [1-2] ;日本界定罕见病为患病人数少于5万(或发病人口比例为1/2500)的疾病 [3] ;欧盟则将其定义为患病率低于1/2000的疾病 [4] 。美国于1983 年颁布世界上首个《孤儿药法案》(The Orphan Drug Act,ODA),极大地激励了美国孤儿药的研发和生产 [5] 。本研究根据FDA孤儿药认定和批准在线数据库和Drugs@FDA数据库,对美国自1983年至2020年认定和批准的孤儿药进行统计分析,为医药企业、医药管理和新药研发部门提供实证参考。
1 数据来源和方法
本研究以美国食品药品监督管理局(FDA)数据库Drugs@ FDA [6] 和FDA孤儿药认定和批准在线数据库 [7] 为主要数据来源,收集整理美国FDA在1983-2020年间通过孤儿药资格认定和批准上市的孤儿药。提取通用名/活性成分(GenericName)、商品名(Trade Name)、资格认定日期(Date Designated)、孤儿药认定状态(Orphan Designation Status)、批准上市日期(Marketing Approval Date)、获批适应症(Approved Labele dIndication)、市场独占期结束日期(Exclusivity End Date)、研发企业(Sponsor)等信息。收集整理美国FDA的药品审评和研究中心(Centerfor Drug Evaluation and Research,CDER)在1985至2020年间批准的新分子实体(New Molecular Entities, NME)信息,包括经新药申请(New Drug Application,NDA)批准的化学新药与经生物制品许可申请(Biologics License Application,BLA)批准的生物新药 [8] 。
根据世界卫生组织(WHO)推荐的解剖-治疗-化学的药物分类体系(The Anatomical Therapeutic Chemical Classification System,ATC分类体系)对药物的治疗领域进行分类;若一种药物具有多个ATC分类编码,则根据FDA批准的新药适应症确定最接近的ATC编码;对于未列入ATC分类的药物,将通过查阅DrugBank、Wikipedia百科或共识,参考FDA批准适应症确定其ATC分类编码。
使用Excel 2019对数据进行描述性统计,分析1983-2020年美国FDA通过孤儿药资格认定及批准上市的孤儿药的特点。
2 研究结果
2.1 FDA资格认定及批准上市的孤儿药数量与趋势
自1983年《孤儿药法案》正式颁布实施以来,获得FDA授予孤儿药资格认定的项目数量和批准的孤儿药适应症数量逐年递增(见图1),截至2020年12月,FDA共授予5757个孤儿药资格,平均每年约为151.5个。1983-2000年,这18年间孤儿药认定数目相对稳定,平均每年约为59.2个,但2001-2020年,这20年间认定数目逐年递增,达到平均每年234.6个。在2017年孤儿药资格认定的数量达到1983年以来的最高值486个,是1984年的约12.5倍。
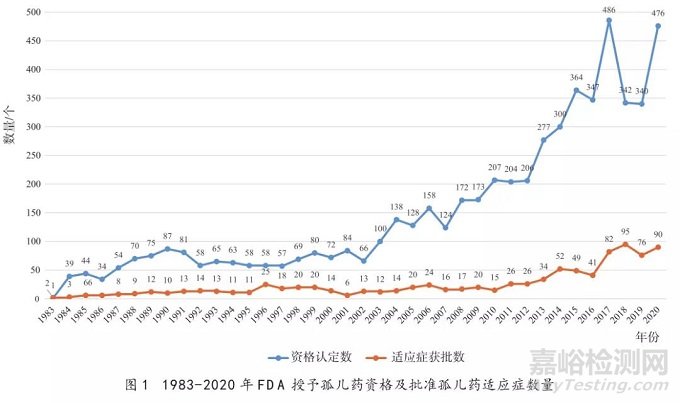
1983年以前,美国上市的孤儿药数量只有10个左右,1983-2020年,FDA共批准上市943个孤儿药适应症(涵盖559种药物,其中有180个药物获批≥2个孤儿药适应症),平均每年约为24.8个。每年批准孤儿药适应症数量整体也呈上升趋势,在2018年达到最高值95个。
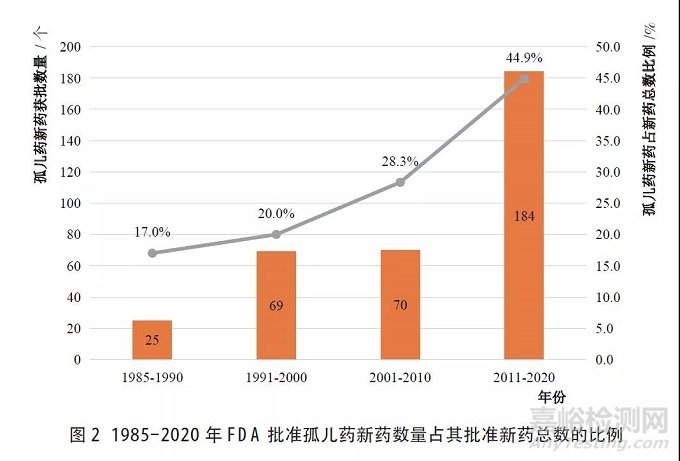
整体来看,1985-2020年FDA批准孤儿药新药数量占其批准新药总数的比例呈增长趋势(见图2),1985年FDA共批准上市31个NME,其中,孤儿药为3个,占9.7%,而2020年,FDA共批准上市53个NME,其中,孤儿药为31个,占58.5%, 占比较1985年明显增大。
2.2 FDA从孤儿药资格认定到获批上市所需的时间
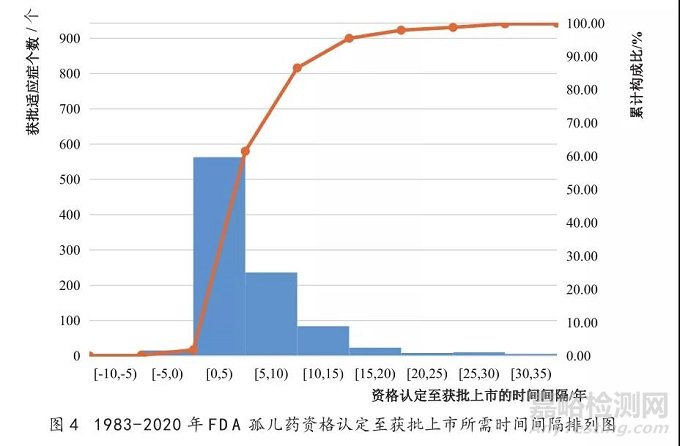
1983-2020年,从孤儿药资格认定到获批上市平均所需的时间约为5.14年。1983-1990年间间隔时间最短,平均约为1.86年,但在之后的3个10年间,间隔时间增加至平均约4.25年到5.94年(见图3)。61.5%的孤儿药适应症从资格认定到获批上市所需时间间隔少于5年,86.5%的适应症所需时间间隔少于10年(见图4)。

2.3 获批上市孤儿药的适应症情况
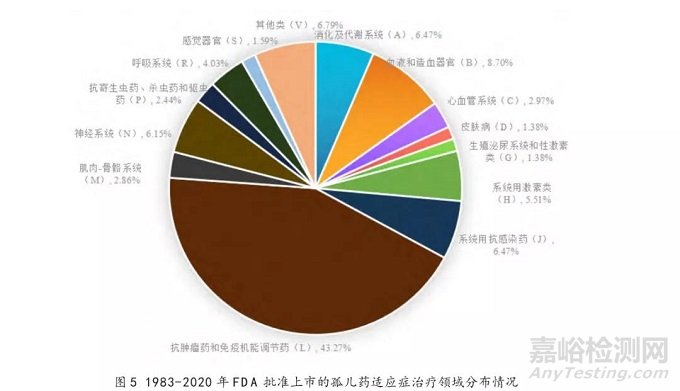
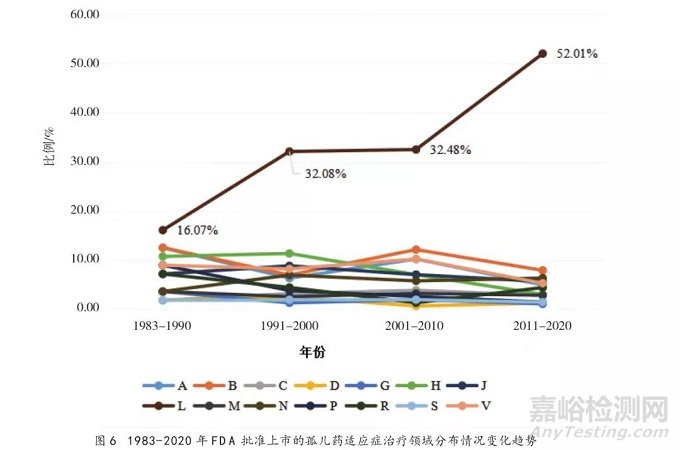
1983-2020年,FDA批准上市的943个孤儿药适应症覆盖多个治疗领域,分布情况具体见表1和图5,其中抗肿瘤药和免疫机能调节药(ATC编码首位L)相关适应症有408个,占比最高,约为43.27%,且呈逐年递增的趋势,由1983-1990年平均占比16.07%,增长至2011-2020年平均占比52.01%(见图6)。


3 讨论
3.1 美国的孤儿药资格与孤儿药批准数量显著增长
在过去的30多年中,FDA授予的孤儿药资格和获批总数大幅增加。近5年FDA批准上市的新分子实体中,孤儿药占了很大的比例。这是多方面因素综合促成的结果:(1)美国的激励政策。1983年1月颁布了《孤儿药法案》,并于1984、1985、1988、2017年进行过4次修订。法案规定,通过孤儿药资格认定后可以享受一系列激励政策:临床研究费用享受50%税收抵免、免除NDA/BLA申请费、可获得专项研发基金资助、特殊的批准通道、免除部分临床数据的申报,以及药物获批后享有7年市场独占期等 [1] 。1992年颁布、2003年修订的《孤儿药条例》(Orphan Drug Regulation)为《孤儿药法案》进一步明确了实施规则。2002年的《罕见病法案》(Rare Diseases Act of 2002)为罕见病研究提供了研究基金支持和法律保障 [9] 。2017年6月,FDA发布“孤儿药现代化计划”(FDA's Orphan Drug Modernization Plan),提出在90天内处理所有提交时间超过120天的孤儿药资格申请,且此后所有新申请须在90天内给予回复,计划刺激了业界申请孤儿药资格的热情 [10] 。(2)技术因素。80%的罕见病都是遗传性疾病,基因组学和基因测序技术的进步,遗传学、生物科学技术等领域的发展,让罕见病药物的识别变得更加精准,制药企业的研发能力显著提高。在批准的孤儿药中有越来越多的生物制剂、罕见癌症治疗药物以及靶向治疗药物。(3)市场因素。高定价及市场独占权等商业驱动因素给研发生产企业带来了高额利润,也刺激了孤儿药开发和生产积极性 [11] 。Hughes研究 [12] 表明,拥有孤儿药上市许可证的公司投资回报率比其他公司高9.6%。Evaluate Pharma发布的《2019年孤儿药报告》(Evaluate Pharma Orphan Drug Report 2019)显示,在美国,孤儿药产品能够获得利润丰厚的定价溢价,孤儿药治疗成本是非孤儿药的4.5倍 [13] 。美国的法案取得了积极的效果,但孤儿药的高定价问题和妨碍市场竞争造成的垄断问题也引起了广泛讨论和争议。
3.2 从孤儿药资格认定到获批上市平均所需的时间呈增长趋势
美国对于孤儿药的注册采取“先资格认定,再上市审批”的方式(简称两步式),相比普通药品,多一步资格认定的前置过程。获得资格认定意味着该药品对于治疗某种罕见病的潜力得到了FDA的认可,可享受研发资助、税收减免、市场垄断等倾斜性政策。从孤儿药资格认定到上市的时间间隔变长,受各种因素影响,其中可能与资格申请的时间以及临床研究的时间有关。尽早获得孤儿药资格认定可以获得更多的融资机会以及在申报审批时可享受美国《孤儿药法案》中的一系列政策优惠。例如,税收优惠是孤儿药法案中重要的优待政策之一,但只对获得孤儿药认定和批准这段时间内的临床试验花费有效,所以在临床研究越早阶段获得资格认定对企业越有利。企业可以在递交NDA/BLA之前的任何时间提交孤儿药资格认定(Orphan Drug Designation,ODD)申请。FDA对孤儿药资格认定的评审重点,一是科学基础,一是影响人口。在涉及ODD申请的科学基础上,由于申请不需要临床数据,所以可使用大量的支持性数据。对2009年获批的160个孤儿药资格认定研究表明,31.88%的申请仅引用了动物试验数据,无临床数据 [14] 。另有数据显示,新药开发正在变得越来越困难和昂贵,临床研究的时间显著增长 [15-16] 。
4 结语
自1983年《孤儿药法案》实施以来,FDA授权的孤儿药资格认定数量和批准的孤儿药适应症数量逐年递增。美国近年来孤儿药快速发展是有多种原因的,其中很重要的一点就是法律和政策的激励。在“健康中国”战略的指导下,我国罕见病防治工作越来越受到重视。2016年2月,原国家食品药品监督管理总局发布《关于解决药品注册申请积压实行优先审评审批的意见》,对包括罕见病治疗药品在内的18种情形的药品注册申请实施优先审评审批。2020年,新修订的《药品注册管理办法》中加快上市注册程序也将具有明显临床价值的防治罕见病的创新药和改良型新药纳入到优先审评审批程序。2018年5月,国家5部门联合制定了《第一批罕见病目录》,收录121个病种。2019年2月,4部门联合发布《关于罕见病药品增值税政策的通知》,对罕见病药品实施税收优惠,对首批21个品种的罕见病药品按照简易办法依照3%征收率计算缴纳增值税。罕见病药物保障政策涉及从药物研发到上市使用的各个环节,需要多部门全方位共同协作的政策保障 [17] 。因此提出如下建议:第一,围绕罕见病的基础研究、药品研发、生产和医疗保障多方面进行顶层设计,借鉴其他国家和地区的激励措施及相关政策,结合我国国情制定“罕见病国家行动计划”。第二,在国家已经出台加速审评审批、提供税收优惠等一系列政策的基础上,加快罕见病立法,从法律层面激励罕见病治疗药物研发,保障罕见病患者权益。第三,健全罕见病药品注册审评机制。借鉴美国经验,罕见病药品注册环节采取资格认定与注册相结合的形式,由国家药品监管机构成立专门的罕见病药品认定部门,组建专门的审评队伍,提高审评效率。第四,加大政府科技投入,尽快设立罕见病领域的重点攻关项目,推动罕见病发病机制和治疗药物的研究,将符合条件、临床急需的罕见病用药列入鼓励药品清单,通过国家科技计划予以扶持和引导。第五,加快建立罕见病治疗药品数据保护制度,形成与专利保护制度相互补充的双重知识产权保护体系。第六,通过严格的专家评审,逐步将疗效确切、医保基金能够承担的罕见病治疗药物纳入医保支付范围。在此基础上,通过建立罕见病专项基金、政策性商业保险、医疗救助等方式实现对罕见病高值药物的医疗保障。通过谈判和集采,将成本降到合理的价位。

来源:中国药房


