您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-02-23 15:14
生物等效性(bioequivalence,BE)研究在创新药物研发、药品上市后评价以及仿制药一致性评价中具有重要作用。
吸入给药是防治哮喘、慢性阻塞性肺疾病等呼吸道疾病的首选给药方式,但由于吸入剂的剂型、装置及作用方式较普通制剂具有特别之处,吸入剂仿制产品的生物等效性研究一般难度较大。
本文主要对比介绍美国FDA、欧洲EMA以及加拿大卫生局(Health Canada,HC)关于吸入制剂(包括经口吸入制剂及鼻喷剂)生物等效性研究的评价方法以及CDE发布的《经口吸入制剂仿制药药学和人体生物等效性研究指导原则》征求意见稿,并结合具体实例进行要点难点浅析,并简要介绍澳大利亚医疗用品管理局(Therapeutic Goods Administration,TGA)的生物等效性评价方法,以期为国内相关吸入制剂生物等效性研究及政策法规的发展提供科学参考。
吸入剂生物等效性研究的评价方法
美国FDA评价方法
FDA建议使用证据权重法进行吸入制剂产品(inhaled drug products,IDP)的BE研究。
这种方法在适当的体外研究、药动学(PK)研究和药效学(PD,或临床疗效)研究所得所有数据的基础上判断给药的等效性。在确保等效性以及患者对替换药物的依从性同时,也考虑处方和给药装置的相似性,见图1。
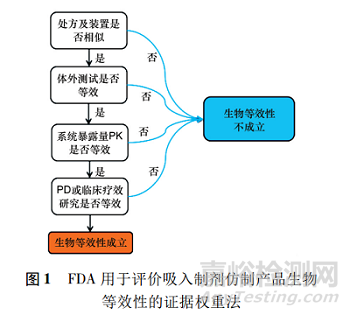
要得出两制剂生物等效的结论,必须同时满足处方及给药装置相似、体外测试等效、系统暴露量PK研究等效以及PD和临床研究等效4个要点,否则生物等效不成立。
欧洲EMA评价方法
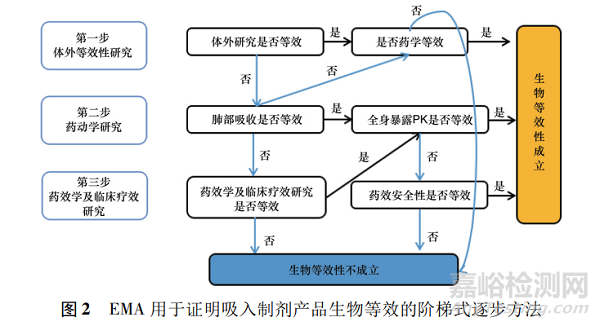
EMA指南使用逐步评估的方法进行吸入剂的生物等效性评价。该方法将体外研究作为第一步,若体外研究结果符合特定等效标准则可直接判定两制剂生物等效;若体外研究结果不满足生物等效标准,则需要进行药动学研究,包括对肺和全身生物利用度的评估。
若药动学研究结果仍不满足等效标准,则第三步使用药效学和临床终点研究证明局部生物等效性(见图2);若可通过药动学研究证明生物等效性,则不需要进一步进行大规模的药效学及临床疗效研究。
加拿大HC和澳大利亚TGA评价方法
HC虽然未在指南中明确表示所用评价方法的类型,但其采取的方法类似于FDA所用的证据权重法:要求使用体外分析及PK,PD研究的所有数据进行生物等效性判断。
TGA在生物等效性评价的某些研究中参考了EMA及FDA发布的指南(主要参考EMA),其评价方法对于不同种类的吸入药物有所不同,但主要采用逐步评估的方法,如定量吸入药物的生物等效性评价,因TGA的大部分指导原则直接引用EMA发布的指南要求,故在下文不再详细描述。
我国CDE评价方法
CDE发布的征求意见稿中亦未表明所用评价方法的类型,其方法参考国家药品监督管理局(National Medical Products Administration,NMPA)发布的相关研究技术及统计学指导原则以及FDA和EMA发布的相关指南,通过判断是否同时满足药学研究等效及人体生物等效(PK,PD及临床研究等效)来判定两制剂的生物等效性,类似于FDA的证据权重法。
吸入剂生物等效性研究
药学研究评价
1、参比制剂的选择
FDA和HC建议从本国的参比制剂(reference listed drug,RLD)中选择,如FDA出版的“批准用于评估生物等效性的药品”(橙皮书),若是使用国外参比制剂需证明该参比制剂与国内参比制剂完全相同,相似的参比制剂不予接受。
EMA则提出只要是通过指定机构审核的原研药品即可作为参比制剂。NMPA在指南中指出为保证仿制药质量,仿制药生物等效性试验应尽可能选择原研产品作为参比制剂。
2、体外生物等效性研究
由于吸入制剂的特点,体外研究所得的生物等效性证据较稳定且对于制剂之间的差异敏感,因此,体外研究对于吸入制剂生物等效性的判断具有至关重要的作用。
在EMA指南中体外测试结果可单独作为生物等效性判断的依据。在其他国家,当无法进行体内药动学或药效学研究时,体外测试结果也可作为生物等效的一个重要附加证据。
体外研究不仅需要评估受试及参比制剂成分及装置的相似性,还需要进行药物递送剂量和空气动力学粒径分布的比较研究。因为这些因素都会影响药物的空气动力学性能,从而影响药物在肺部的沉积量,进而影响药物的临床疗效。
例如,Singh等分别在对一种新的噻托溴铵干粉吸入剂PUR0200进行药效学及安全性评估时,发现当受试制剂与参比制剂相比其细颗粒所占比重更大时,受试制剂中的有效成分在肺部沉积的比例更高,其系统暴露量更低,临床疗效及安全性更好。
Perry等还得出吸入制剂的药动学及疗效等效性可通过微粒粒径大小来初步预测的结论。
绝大多数文献中在进行两制剂生物等效性判断的体外研究时均进行成分及装置、递送剂量的均匀性及空气动力学颗粒大小的研究来评估体外等效性,如Srichana等及Ammari等。
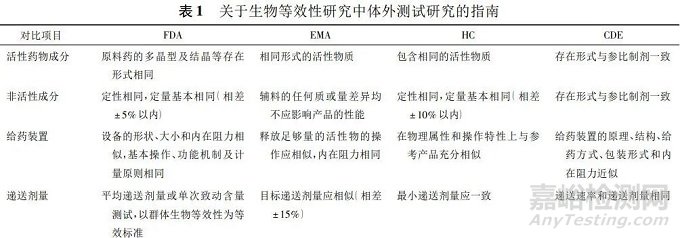
勃林格殷格翰公司在对一种包含噻托溴铵和沙美特罗的新型干粉吸入剂进行体外测试研究时,按照EMA指南进行比较研究发现,受试及参比制剂不仅药物成分和装置相似,而且级联撞击物质量和空气动力学研究的细颗粒质量的差异均在可接受范围内(±15%),故依次判断两制剂在体外等效。表1总结了国际及国内对生物等效性研究中体外测试研究的指南建议。

人体内生物等效性研究
吸入剂生物等效性的人体内研究包括PK、PD和临床终点研究。
FDA于2019批准上市的舒利迭(Advair Diskus)的首款仿制制剂氟替卡松/沙美特罗干粉吸入剂(Wixela Inhub)就分别于健康受试者及哮喘患者体内进行PK及PD研究,PK判断标准为浓度-时间曲线下面积(AUC)和血浆峰浓度(Cmax)2个指标受试与参比制剂的几何均值比的90%置信区间是否在FDA指定的生物等效范围内,PD主要指标为第1秒用力呼气容积(FEV1)。
人体PK研究的主要目的是测定吸入药物活性分子的全身暴露量,以监测受试制剂的安全性。例如,在前文提到的Singh等在对一种新的噻托溴铵干粉吸入剂PUR0200进行安全性评估时,发现受试制剂的系统暴露量更低,不良反应的发生率也更低,即安全性更好。
Algorta等在健康志愿者体内研究一种新型噻托溴铵单糖胶囊干粉吸入制剂的BE时也是通过测定血浆中药物含量分析全身暴露量水平以判断制剂的安全性是否等效。
PD和临床终点研究则通常用于评估局部作用的临床效果,根据主要及次要指标数据来判断疗效是否等效。例如,Horhota等在对一种新型干粉吸入剂进行药效学研究时,采用FEV1作为主要PD指标来判断受试及参比制剂的局部疗效等效性。以下就这3种研究方法进行简要介绍与对比。
1、人类PK-BE研究
由于FDA,HC和CDE要求最终等效性结果的判定需结合体内体外所有过程所得数据,因此对吸入制剂进行PK研究的目的仅为比较受试制剂与参比制剂的全身暴露量和安全性。
而EMA可单纯通过PK研究得出等效结论,因此其PK研究等效所需达到的标准较高,且研究目的也不仅限于全身暴露及安全性研究,还包括在排除胃肠道中活性成分吸收的前提下对肺部沉积量的比较。
Omar等指出,PK研究对受试制剂和参比制剂在药物递送方面的微小差异十分敏感,因此适合评估生物等效性。PK生物等效性参数的90%置信区间的可接受范围(80.00%~125.00%)反映了适当的保守范围,以确保受试制剂和参比制剂的安全性和有效性相同。

表2是国际及国内关于人体PK研究的指南对比。
苯丙米松二丙酸酯/富马酸福莫特罗加压定量吸入剂、沙美特罗/丙酸氟替卡松定量吸入剂、布地奈德/福莫特罗干粉吸入剂、沙丁胺醇气雾剂等吸入制剂均进行了体内PK研究,通过对PK参数AUC及Cmax的统计分析,判断受试制剂及参比制剂在PK方面的等效性以支持药物的生物等效性评价。
2、人体PD-BE及临床终点研究
在各生物等效性研究指南中都提到了使用PD及临床终点研究的方法,该方法应包括证明受试及参比制剂疗效等效及系统安全性等效。例如,Kuna等在对苯丙米松二丙酸酯/富马酸福莫特罗加压定量吸入剂在30例患有哮喘的青少年体内的PD研究中,不仅检测两组间的血浆钾、葡萄糖、脉搏和肺功能等PD变量以判断疗效等效性,还测定受试者血浆中药物有效成分的含量以计算PK参数,用于判断药物的系统暴露量,即安全性是否等效。
Singh等在对一种新的噻托溴铵干粉吸入剂PUR0200与参比制剂噻托溴铵HH的生物等效性研究中,不仅研究主要PD变量:FEV1的曲线下面积以判断疗效等效性,还研究计算PK参数,以判断两制剂的全身暴露量是否等效。
Bhattacharya等在证明一种新的含氢氟烷的压力定量吸入剂对于治疗慢性阻塞性肺疾病与传统氯氟碳压力定量吸入制剂相比是临床非劣效的研究中,亦在评价主要PD变量(FEV1)变化的同时进行药物的全身暴露评价。表3是国际及国内关于人体PD及临床终点研究的指南对比。

目前市面上许多吸入制剂是由糖皮质激素类药物组成的,在对这些含有激素类药物进行生物等效性评价时,需要同时监测激素对下丘脑-垂体-肾上腺(hypothalamic-pituitary-adrenal,HPA)轴的影响,以证明受试制剂对HPA轴的影响不大于参比制剂。
除以上几种PD指标外,在特殊的情况下也可使用经证明更加敏感有效的疗效指标。例如,有研究表明用乙酰甲胆碱进行支气管激发,FEV1作为药效学指标是测定吸入型支气管扩张剂BE最敏感的方法。但许多儿童不能进行重复性的肺活量测定。
Mondal等旨在研究以脉冲振荡技术所得数据为PD指标时,乙酰甲胆碱激发是否能区分2种沙美特罗吸入剂。试验对象为10例4~11岁的轻度稳定型哮喘患者,在吸入受试制剂或参考制剂后进行乙酰甲胆碱激发,计算引起气道总阻力增加40%时的乙酰甲胆碱激发浓度。结果显示以脉冲振荡技术所得数据作为PD指标也是一种确定儿童用吸入剂生物等效性的有效方法。
吸入剂生物等效性研究要点难点浅析
体外研究
吸入制剂体外研究方法一般较严谨,数据稳定,对制剂之间的差异也比较敏感,但药物在体内外经历的过程不完全相同,因此药物体外研究的结果不能够完全代表药物的体内过程。
Horhota等对包含噻托溴铵和沙美特罗的一种新型干粉吸入剂的体外测试研究发现,其级联撞击物质量和空气动力学细颗粒质量均与参比制剂相似:差异<15%,符合体外生物等效条件,体内PD指标也未发现显著性差异。然而在体内PK研究中由于受试制剂的尿排泄量较高,AUC及Cmax等指标存在显著性差异,PK研究不等效,表明该吸入制剂的体外结果并不能很好地预测临床PK发现。
因此,建立体内体外-药物相关性(in vitro-in vivo correlation,IVIVC)的评价方法,对于体外测试更好地预测体内结果是至关重要的。
EMA关于修订口服吸入产品临床要求指南的概念文件中也提到了IVIVC研究数据对于BE研究的重要性。Lahelma等正是凭借IVIVC建模的研究预测结果以及多项PK研究结果,使当时新开发的一种包含布地奈德和福莫特罗富马酸酯二水合物的干粉吸入剂在欧洲获得批准,用于替代信必可都保治疗哮喘和慢性阻塞性肺疾病。
对于吸入制剂IVIVC评价方法的开发也吸引了许多研究者的兴趣,例如,Weers等在建立气雾剂性能与PK和PD之间IVIVC的研究表明,在测试条件下,真实喉道模型可在肺沉积及药物系统暴露水平方面提供良好的IVIVC(与体内平均测量值的差异为5%~15%)。
FDA仿制药物办公室(the Office of Generic Drugs,OGD)也在进行将体外测试与计算机技术相结合以更好预测药物体内过程的技术开发:FDA在2016年对局部作用的口服吸入和鼻用药物产品的监管科学报告中表示OGD正在开发计算流体动力学(computational fluid dynamic,CFD)和基于生理的药动学(physiology-based pharmacokinetic,PBPK)模型,以预测吸入制剂在人体内的过程,并评估其在仿制药开发计划中的适用性。
因此,继续开发合理的IVIVC评价方法,对于更好地开展及规范吸入制剂BE评价,以及降低成本、提高时间效益具有深远的意义。
体内PK研究
对于发挥全身作用的一般口服制剂,在给予受试制剂或参比制剂后二者的PK参数之间的差异在可接受范围内,通常即可认定生物等效。但吸入制剂在进入体内后,首先分布到作用部位,再进入体循环,同时还通过其他部位(如口、咽、胃肠道等)进入体循环,药动学和局部递药等效性之间关系复杂。
在EMA指南中提到PK研究目的有2个,一是评估肺部沉积,二是研究全身暴露量以评价安全性。
当PK研究用于评估肺部沉积时,应排除从胃肠道吸收活性成分,在EMA口服吸入产品指南中,要求将与疗效相关的肺部暴露与口服吸入产品施用后发生的总全身暴露区分开来。
区分肺部接触和非肺部接触的方法包括荧光显像法和口服木炭阻滞法。目前,荧光显像法还没有被接受作为确定生物等效性的方法,主要是因为参考制剂的放射标记十分复杂。而口服木炭阻滞法在评估口服吸入产品时将肺部暴露与非肺部暴露区分开有很大的优势,已经被广泛应用,并且已被写入EMA指南。
在使用各种方法排除活性成分被吸收时需保证并证明数据的准确性。除评估肺部沉积外,国际上对于PK研究都规定需评估全身暴露量以评价药物安全性。对于全身暴露量评估涉及的研究剂量、受试者的选择等,都应特别注意。
FDA规定用于吸入剂PK研究的药物剂量应能通过灵敏的分析检测方法得出PK研究曲线。对于分析检测方法的确定及最终药物剂量的确定都是PK研究中需要仔细考量的难点。FDA有关研究表明,健康受试者的PK数据通常比患者体内的PK数据要稳定,这可能与患者体内和疾病相关的各种变化有关。因此FDA对于吸入剂PK研究的受试者要求健康成年受试者,以期得到关于受试制剂及参比制剂差异的更稳定数据以判断等效性。
体内PD及临床终点研究
PD指标的选择应该是能够最好地显示药物作用效果的单一指标或复合指标。在PD及临床疗效研究中,药物剂量、PD指标的选择对确定指标灵敏的测定方法以及评价标准均对试验有十分重大的影响。
因此,对于这些指标及方法等的选择也是生物等效性研究中的一大难点,需要通过对大量数据及报告或试验、实验的研究以最终确定有效指标等。例如,在对哮喘控制的评估中,哮喘状态以及结局指标等的确定对于临床疗效研究至关重要,Devadason等分成多个研究小组通过阅读研究近500篇文献最终确定用于哮喘控制评估的复合指标。
总结
本文对美国、欧盟、加拿大和我国吸入剂生物等效性研究指南的异同进行综述。
生物等效性研究主要包括体外测试、体内PK研究以及PD和临床终点研究3个阶段,但各阶段的优先度以及设计方法等各国的侧重点有所不同。
吸入剂生物等效性研究设计的指南十分重要,Zeng等通过对FDA关于沙丁胺醇生物等效性评价的指南设计进行改进,发现当研究剂量进行合理的改变时,可以使样本量和研究成本得到有效降低。因此我们需要了解更多地区的相关指南来丰富我们对吸入制剂生物等效性评价的认识,以期能得到更高效的指南为国内吸入制剂的发展助力。
考虑到吸入剂的特点,吸入剂体外、PK及PD研究有其特殊性,但大多数指标生物等效性标准仍与一般药物要求的主要PK参数的T/R几何均值比90%置信区间为80.00%~125.00%基本相同。
在进行吸入剂生物等效性研究时,既要考虑其特殊性以保证试验数据及结论的准确,也要思考其与一般制剂的共性,更好地规范试验研究设计。
目前各国对于吸入制剂生物等效性评价的研究仍在不断改进更新,越来越多的体内或体外模型、IVIVC及PBPK技术等用于完善研究,以期能以更低的成本、更高的效率及准确度完成生物等效性评价。完善我国发布的征求意见稿时,在参考目前各国发布的指南的同时也应参考现阶段经过验证的各种新型辅助研究技术,以更好地促进及规范我国吸入制剂的生物等效性研究。

来源:中国新药杂志


