您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-04-04 18:00
摘要
无定形固体分散体是提高难溶性药物生物利用度最有效的策略之一,但其易受到处方因素、制备工艺、存储条件和溶出条件等因素的影响从而在储存期或溶出过程中结晶, 丧失溶出优势。此外, 体内外环境的差异、表观浓度与透膜通量之间的差异、体内吸收过程的复杂性等影响因素使得无定形固体分散体的体外溶出不能完全准确预测体内吸收, 给固体分散体产品开发带来了极大挑战。
本文总结了关于无定形药物固体分散体溶出与吸收的研究进展, 期望为难溶性药物无定形固体分散体制剂的开发提供参考。
关键词
无定形固体分散体; 难溶性药物; 结晶; 体外溶出; 体内吸收
正文
随着分子设计、高通量筛选与组合化学、机器人自动合成技术的发展, 涌现出越来越多低溶解度的候选药物[1-3]。候选化合物较低的溶解度往往会导致较低的口服生物利用度, 给成药带来巨大的挑战。有众多的制剂学策略已用于提高难溶性药物的溶解度, 如前药、自乳化系统、固体分散体、纳米晶、共晶、环糊精包合物、固体脂质纳米粒、脂质体及胶束等技术[4-7]。其中, 无定形固体分散体(amorphous solid dispersion, ASD) 技术由于增溶效果好[8]、安全性高[9]、血药浓度波动小[4]、易于工业化生产[10]等优势, 已成为难溶性药物增溶的重要手段之一。
ASD 的溶出优势归因于药物无定形化和形成固体分散体。药物无定形化后,药物处于高能态, 溶出时释放的能量更大, 促进了溶出, 即图1 中Ec > Ed[6]; 同时,溶出时无定形药物无需克服晶态药物溶出时所需的晶格能, 加快了溶出, 即图1中Ea < Eb[4,7]。
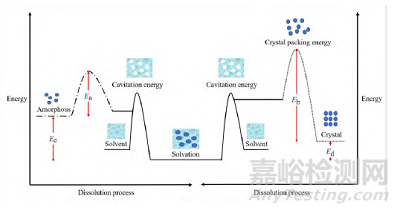
Figure 1 The energy in dissolution processes of amorphous andcrystalline drugs, Ea and Eb are the energy barriers during the dissolution of theamorphous and crystalline states, respectively; Ec and Ed are the released energy duringthe dissolution of the amorphous and crystalline states, respectively
此外, 固体分散体增溶的机制还包括润湿性提高[5]、去过饱和与结晶行为受到抑制[8,11]、溶出时所需空化能减小[4,12]、表面活性剂增溶作用[5]等。综上, 药物无定形化和分子层面的分散特性共同促进了ASD 的溶出行为, 使得ASD 技术成为提高难溶性药物溶解度最有效的方法之一。
虽然 ASD 技术具有显著的增溶效果, 但无定形药物溶出时可能受多种因素影响而导致结晶, 溶出过程中可能伴随发生多种物理变化, 如液-液相分离[13-16]、表面活性剂的增溶作用[17,18]、无定形药物表面结晶[11,19]和表面相分离[20]以及药物过饱和溶液的去过饱和[21,22]等现象, 进而影响ASD 的溶出行为。上述物理变化中最为常见的现象为过饱和溶液的去过饱和以及无定形制剂的表面结晶。去过饱和是指ASD 溶出形成过饱和溶液, 易析出晶体从而导致溶液中药物浓度降低的现象[8]; 表面结晶是指无定形药物表面存在的一种速率更快的结晶行为[11,19]。
由于无定形药物溶出时易发生结晶, 导致过饱和溶液中药物的浓度降低, 因此添加抑制结晶的辅料可有效缓解上述问题。根据辅料对结晶的抑制效果, 无定形药物的溶出曲线可分为弹簧模型或弹簧-降落伞模型。当辅料抑制结晶效果较差时, 药物溶出快速形成过饱和溶液, 但由于快速去过饱和与无定形药物表面结晶等原因, 溶液中药物浓度骤降, 溶出曲线表现为弹簧模型; 如果加入的辅料能有效抑制药物溶出过程中的结晶, 从而延长过饱和溶液的状态, 此时溶出曲线为弹簧-降落伞模型[23]。
此外, ASD 的体内溶出和吸收过程会受到生理环境和给药方案等因素的影响。Newman 等[24]的统计表明, 在报道的ASD 案例中, 有8%的ASD 生物利用度反而降低。因此明确ASD 体外溶出和体内吸收的相关影响因素及内在机制具有重要意义。本文首先介绍ASD 具有溶出优势的相关机制, 及ASD 溶出的各种影响因素, 阐述现阶段通过ASD 溶出结果预测体内吸收面临的挑战, 最后介绍近年来ASD 制剂研发的相关进展, 旨在为ASD 产品开发提供借鉴和参考。
1 ASD 体外溶出的影响因素
1.1 处方因素
1.1.1 药物自身性质
药物无定形态与晶态之间的能量差异、无定形态的物理稳定性和成胶性对ASD 溶出具有较大影响。首先, 药物无定形态与晶态间的能量差异会影响其无定形的增溶效果, 不同药物的无定形溶出优势不同, 即使结构类似物也可能存在显著差异。如相比于利托那韦, 阿扎那韦的无定形态与晶态之间能量差异更大,其无定形溶出优势也更显著[25]。其次, 不同药物的无定形态自身结晶趋势不同,结晶趋势较强的无定形药物在储存期间结晶风险较大, 一旦ASD 发生结晶, 溶出优势将降低。Edueng 等[26]研究发现, 无定形酮康唑在25 ℃/75% 相对湿度储存2天后结晶度达到23%, 体外溶出降低了50%, 溶出时最大浓度与晶态溶解度相当。
结晶速率快的药物在溶出时往往固体表面结晶速率及溶液去过饱和速率均较快,从而降低溶出。此外, 某些无定形药物接触溶出介质后, 自身形成胶状物质对ASD 溶出也有一定影响。如无定形盐酸鲁拉西酮接触溶出介质后发生凝胶化,导致其溶出度低于晶态[27]。综上, 选择无定形态与晶态能量差异较大、结晶趋势较弱、结晶速率较慢的药物, 更有利于ASD 制剂的开发。这些性质分别与药物的结构、熔点和熔化焓[25]、玻璃化转变温度[28]等因素有一定相关性。药物自身理化性质对ASD 溶出行为影响显著, 因此处方开发前需对药物的理化性质进行全面分析。
1.1.2 载药量
ASD 载药量不同有可能改变ASD 的溶出机制。根据高分子和药物释放速率的相对快慢, ASD 溶出机制分为3 种 (图2)[29]。
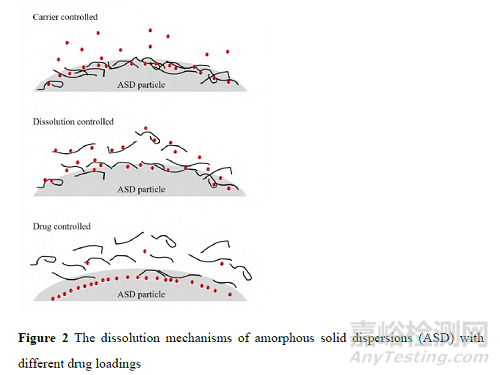
当高分子释放速率较慢时, 药物需通过高分子层扩散到溶液主体, 此时溶出行为受载体控制 (carrier controlled);当药物和高分子同步快速释放时, 能产生过饱和溶液, 此时溶出机制为溶出控制释放 (dissolution controlled); 当高分子释放速率较快时, ASD 颗粒表面药物含量提高, 溶出行为受药物控制 (drug controlled), 此时如果无定形药物的物理稳定性较差, 可能会诱发表面结晶, 从而阻止后续药物溶出, 难以继续生成并维持过饱和溶液[30]。当ASD 的载药量发生变化, 可能导致上述溶出机制的改变, 从而影响溶出。Que 等[20]研究表明, 当载药量超过临界值时, ASD 的溶出机制主要为药物控制释放, 该作者认为这是由于高载药量的ASD 溶出时发生了表面相分离, 在片剂表面上形成了富药相而阻碍了片剂内部物质的进一步溶出。从临床顺应性的角度来看, 高载药量的制剂通常会有更多的优势。但ASD 开发过程中应充分研究和评估高载药量制剂可能带来的溶出结晶风险。
1.1.3 高分子辅料
高分子聚合物作为 ASD 的载体材料, 对药物溶出释放往往具有决定性影响,不仅可抑制无定形态药物结晶, 同时还能维持药物的过饱和状态。高分子有多种不同分类方式, 对于药用高分子, 常根据单体或溶解性进行分类。按单体分类, 可分为聚维酮类、聚乙二醇类、纤维素类、聚丙烯酸类等; 按溶解性分类, 可分为无pH 依赖型、酸溶型和碱溶型等。由于高分子的溶解性会显著影响ASD 药物释放行为, 因此对ASD 制剂开发中选择高分子具有一定指导意义。高分子影响ASD 溶出的机制可总结为以下几点。
1.1.3.1 高分子降低去过饱和速率
高分子可通过药物-高分子相互作用、增加溶液黏度、晶面吸附等多种机制抑制去过饱和。Amponsah-Efah 等[31]运用超速离心技术, 表明溶液中药物-高分子相互作用越强, 去过饱和行为越弱。Hanada 等[32]研究表明, 虽然高黏度型羟丙甲基纤维素 (hydroxypropyl methylcellulose,HPMC) 与药物的相互作用弱于低黏度型HPMC, 但高黏度型HPMC 增加了溶液黏度和药物结晶时所受的位阻, 因此抑制过饱和溶液析晶的能力更强。此外,Schram 等[22]研究发现, 醋酸羟丙甲纤维素琥珀酸酯 (hydroxypropylmethylcellulose acetate succinate, HPMCAS) 通过吸附在晶面上, 抑制非洛地平过饱和溶液的析晶。
1.1.3.2 高分子包衣抑制溶出表面结晶
高分子可抑制药物表面结晶从而改善ASD 的溶出行为。Li 等[11]通过对无定形吲哚美辛表面进行壳聚糖包衣, 抑制了储存和溶出过程的表面结晶, 从而改善了药物溶出。1.1.3.3 高分子增加了储存期ASD 物理稳定性 ASD 在储存过程中可能会发生结晶, 即使ASD 中含有少量药物晶体, 体外溶出和体内吸收均可能显著降低。当处方设计合理时, ASD 长时间储存后不会因结晶而丧失溶出优势。如硝苯地平-共聚维酮-聚甲丙烯酸铵酯ASD 在室温储存25 年后, 未发生明显结晶或化学降解,溶出行为与新制备的ASD 差异不大[33]。
1.1.3.4 高分子控制药物释放速率
Sun 等[34]研究表明, 不同种类的高分子可调控ASD 释放速率, 从而影响溶出行为。当处方中加入易溶性高分子, ASD 的溶出速率较快, 溶液迅速达到过饱和状态, 易导致体系快速成核与析晶; 相反, 当处方中加入难溶性高分子, ASD 的溶出速率较慢, 体系能在较长时间内维持过饱和度。Ojo 等[35]研究发现, 相较于难溶性高分子, ASD 处方中添加水溶性高分子后药物释放速率更快。但对于残留有晶体的ASD, 药物快速溶出使得残留的药物晶体被释放出来, 易成为晶体生长的位点, 导致ASD 溶出形成的过饱和溶液快速析晶, 从而降低溶出。因此高分子的溶出释放将影响药物的释放速率, 从而改变药物在胃肠道释放过程中过饱和度的形成快慢。
1.1.3.5 高分子控制药物定位释放
高分子可在一定程度上调控ASD 在体内的定位释放。Winslow 等[36]通过将肠溶性高分子作为ASD 载体, 控制药物肠道定位释放, 解决了利福丁在胃部溶出时酸性环境降解的问题, 实现了增加溶解度和溶液中化学稳定性的平衡。
1.1.3.6 高分子影响ASD 的崩解行为
高分子对ASD 崩解的影响也会影响药物的释放。Zhang 等[37]研究发现, ASD 处方中加入疏水性高分子HPMCAS 比加入亲水性高分子乙烯基吡咯烷酮/乙酸乙烯酯共聚物 (polyvinylpyrrolidone vinylacetatecopolymer, PVP/VA) 或HPMC 崩解更快, 从而释放速度更快。该作者认为这是由于亲水性高分子接触水后易于胶化, 导致溶出介质难以进入片剂内部, 阻碍了ASD 片剂的崩解, 从而抑制了药物溶出与释放。
1.1.3.7 高分子影响液-液相分离 (liquid-liquid phase separation, LLPS) 形成
对于结晶较慢的药物, 当溶液中的药物浓度超过无定形溶解度时, 药物以无定形富药相的形式形成纳米粒, 这种现象称为LLPS[29]。无定形富药相又被视为过饱和溶液的储库和结晶的前体, 前者可进一步维持过饱和度从而促进溶出, 而后者将诱导结晶从而降低溶出, 因此该富药相对溶出的利弊取决于药物的结晶速率[13]。Taylor 等[38]根据药物-水不同比例下的混合吉布斯自由能图 (图3), 指出溶液中药物浓度不同时, 形成LLPS 的难易程度不同。
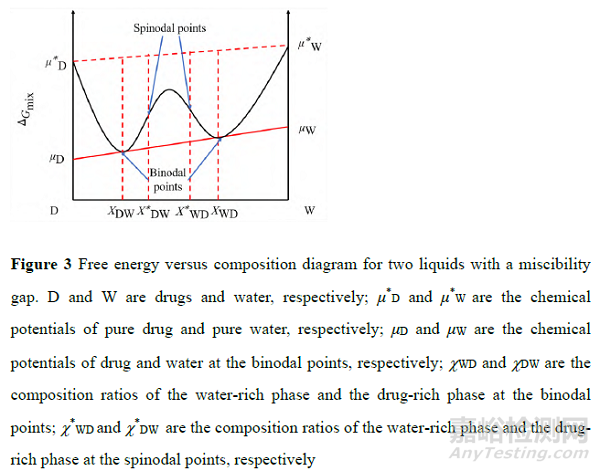
高分子通过稳定LLPS 或降低无定形药物溶解度而影响溶出。多篇相关报道表明, 高分子可通过吸附在药物富药相表面或混入药物富药相中, 抑制相分离液滴的聚集和熟化, 稳定了液-液相分离体系从而维持了药物较高的过饱和度[15,39,40]。Ueda 等[39]将HPMC 抑制布洛芬ASD 溶出时的析晶归因于高分子抑制了药物富药相中药物的运动性, 而非高分子对游离态药物的作用。但高分子也可能混入LLPS 的药物富药相中, 降低药物的化学势从而降低无定形药物溶解度[40]。
通常药物的化学势是指溶液中药物的自由能, 由于该值与透膜通量相关, 所以化学势的降低同时也降低了透膜通量, 不利于体内吸收[29,40,41]。因此, 既能稳定LLPS 又不影响无定形溶解度的高分子能提供更优异的溶出和吸收性质。
因此建议ASD 开发过程中, 应当综合考虑高分子对无定形药物结晶行为、释放动力学以及LLPS 形成的影响, 针对不同ASD 制剂所面临的具体问题, 合理选择高分子辅料。此外, 在充分理解高分子影响ASD 溶出机制的基础上, 可筛选出合适的高分子辅料组合, 以期发挥协同作用提高ASD 的溶出。
1.1.4 表面活性剂
ASD 处方中常加入表面活性剂, 通常可进一步增加药物溶解度, 目前已有相关ASD 产品上市。如治疗囊性纤维化的Orkambi®, 其处方中添加了表面活性剂十二烷基磺酸钠 (sodium lauryl sulfate, SLS)[10]。Wang 等[18]系统研究了不同表面活性剂对硝苯地平过饱和溶液的影响, 结果表明SLS 等6 种表面活性剂对药物的增溶作用和维持过饱和溶液的能力不同, 这两点被认为是表面活性剂影响ASD 溶出所形成的过饱和溶液的主要因素。同时, 还需关注高分子和表面活性剂共同对ASD 溶出的影响。部分高分子与表面活性剂联用时, 如PVP/VA 与牛磺胆酸钠, 可起到协同抑制结晶的作用, 其原因在于表面活性剂与高分子的结合位点不同从而增加吸附面积, 或表面活性剂增加了高分子对药物的亲和性[18]。
但Chen 等[42]研究发现, 由于SLS 围绕HPMCAS 自组装形成胶束, 以及SLS 与HPMCAS 的相互作用减弱了HPMCAS 的抑晶效果, 表面活性剂SLS 的加入反而使含HPMCAS 的ASD 溶出降低。由于越来越多的ASD 处方中添加了外源性或内源性表面活性剂, 表面活性剂对ASD 体外溶出和体内吸收的影响不可忽视, 理解表面活性剂影响ASD 溶出的本质原因有助于设计处方时选择合适的表面活性剂以及预判ASD 体内吸收行为。因此在ASD 开发过程中选择表面活性剂时, 除了考虑其增溶能力外, 建议还需考察表面活性剂的加入对ASD 储存和溶出时物理稳定性的影响。此外, 即使ASD 处方开发过程中不加入表面活性剂, 也应考虑内源性表面活性剂对ASD 制剂体内溶出与吸收的影响。
1.1.5 其他常见辅料
ASD 制剂中除了药物、高分子及表面活性剂外, 还可添加其他功能性辅料。其他功能性辅料可能通过影响ASD 崩解行为从而间接影响ASD 的溶出。如Heng 等[43]通过向处方中添加1%~4%疏水性硅胶Aeroperl® R972, 抑制了溶出时无定形吲哚美辛的胶化行为, 从而改善了溶出。Takano 等[44]的研究也表明, 分别在处方中加入碘化钾、氯化钾等4 种盐后, 仅2%的添加量就能显著改变伊曲康唑-HPMC ASD 的溶出行为。其背后的原因在于盐溶离子抑制了HPMC 的成胶现象, 促进药物释放; 盐析离子对ASD 溶出的影响则恰好相反。此外, Agrawal 等[45]系统研究了崩解剂对ASD 溶出的影响, 结果表明交联聚维酮和交联羧甲基纤维素钠的促进崩解效果最好。另外, 泡腾剂碳酸氢钠可促进ASD 片剂的崩解, 但效果不如无机盐氯化钠[46]。
1.2 制备工艺
ASD 不同加工过程可能会导致ASD 产品质量不同, 进而对后续的溶出造成影响。如对比电纺丝法和喷雾干燥法制备的螺内酯-PVP/VA ASD[47], 后者溶出效果较差, 借助偏光显微镜证实了喷雾干燥制备的ASD 产品有一定的结晶度。该作者认为由于喷雾干燥过程中溶剂蒸发速率慢, 因此在制备ASD 的过程中随着溶剂的缓慢蒸发, 可能诱发了结晶。除了不同工艺, 同种工艺的不同参数也会导致ASD 质量不同, 从而影响溶出。Moseson 等[48]通过控制热熔挤出的加工温度和时间等参数制备了残留不同药物晶体含量的ASD, 结果显示结晶度越高的ASD 在溶出过程中的去过饱和现象越明显。如果热熔挤出工艺条件控制不佳,比卡鲁胺-PVP/VA ASD 内部会残余部分比卡鲁胺稳定晶型, 溶出时所能达到的过饱和度较低, 残余的晶体更易诱发无定形药物的固相结晶, 同时伴随药物浓度下降的去过饱和现象[49]。综上, 不同工艺或同种工艺的不同参数均会造成ASD产品质量和后续释放行为的差异。因此需合理设计相关ASD 工艺,可考虑过程分析技术 (PAT) 等手段,实时监测工艺参数以确保产品质量。此外, 开发新的ASD 制备工艺也是未来的研究方向之一。
1.3 存储条件
由于 ASD 制剂存在运输期和存储期, 不可忽略存储条件影响ASD 物理状态从而影响后续ASD 溶出的可能性。其中, 储存温度和储存湿度对ASD 储存期物理稳定性的影响尤为显著, 当存储期发生结晶, ASD 的溶出优势将降低[28]。Tian等[50]研究发现在不同温度及湿度的加速条件下储存后, ASD 的结晶度不同。进一步研究发现, ASD 中药物的结晶程度与药物溶出释放量呈线性负相关, 而与溶出速率不存在相关性。因此储存条件主要通过诱导无定形药物在储存期间的结晶从而间接影响溶出与吸收。在开发ASD 处方时, 可考虑设计防潮包装, 增加对制剂的保护。其次, 在设计处方时可添加合适的辅料来调节药物的宏观分子运动性, 该指标通常通过药物的玻璃化转变温度衡量。经验性规律表明, 当ASD 制剂的储存温度远低于其玻璃化转变温度 (相差超过50 ℃以上时), 此时无定形药物的分子运动很慢, 通常具有优良的物理稳定性[28]。因此可通过加入抑制无定形药物分子运动的辅料及提高其含量, 提高ASD 制剂的玻璃化转变温度, 从而增加制剂稳定性。
1.4 溶出条件
溶出条件相关的各因素中影响最显著的是溶液 pH 值。pH 通过改变溶解度、影响溶出微环境和离子吸附抑制析晶等显著影响ASD 溶出。与其他普通制剂类似, 溶解度改变假说认为pH 会改变药物晶态和无定形态的溶解度, 进而影响溶出行为。Indulkar 等[14]研究发现, 随着pH 的降低, 克霉唑等3 种弱碱性药物的无定形态和晶态溶解度均增加, 但是两者比值保持不变。微环境pH 差异性假说认为过饱和溶液析晶时, 溶液本体和颗粒表面微环境的pH 不同, 导致去过饱和行为上的差异。Chen 等[51]的研究表明, 加入氢氧化钠后, 吲哚美辛-PVP/VA ASD溶出增加。该作者认为无定形吲哚美辛与氢氧化钠形成了无定形盐, 当这种高溶解度盐释放时, 增加了颗粒溶出时扩散层微环境的pH 值, 从而进一步促进了吲哚美辛的溶出。离子吸附假说认为过饱和溶液析晶时, 析出的晶体在生长过程中其晶面会吸附离子化药物或高分子, 占据非离子化药物结晶生长位点[21,22]。由于不同pH 下晶面吸附程度或吸附构象不同, 从而导致去过饱和行为上的差异。如吲哚美辛在较高pH 时下去过饱和速率更慢, 这是由于吲哚美辛晶面吸附电离的吲哚美辛从而抑制进一步析晶, 因此能维持较高的过饱度[21]。此外, Schram 等[22]研究发现, 不同pH 环境下HPMCAS 对非洛地平过饱和溶液的抑晶作用存在差异, 其原因在于不同pH 下高分子构象不同, 对药物晶面的吸附能力不同。由于pH 对ASD 溶出影响的复杂性及体内吸收过程中pH 的改变, 在设计体外溶出实验条件时, 应充分考虑体内环境的pH 变化情况。如可在溶出前期使用酸性介质,溶出后期改用碱性介质的方法, 更好模拟体内真实情况, 降低体内外差异风险。
ASD 在固态条件下维持稳定, 是其维持溶出优势的前提条件。因此在处方筛选时, 特别是选择高分子载体材料时, 应兼顾ASD 固态条件下的稳定性和快速溶出速率。
内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果,注明来源于凡默谷公众号。
2 ASD 体内吸收
ASD 增溶的最终目的是提高难溶性药物的体内吸收生物利用度。根据Newman 等[24]的统计, 约有82%的研究表明ASD 与原料药相比能提高生物利用度, 且主要通过促进药物溶出从而促进吸收。此外, 抑制P 糖蛋白外排、促渗透作用、胃肠道的生物黏附、磷脂双分子层扰动及防止pH 和代谢带来的降解等因素也可能促进药物吸收[52]。虽然普遍认为ASD 通过改善溶出从而增加生物利用度, 但Newman 等[24]的统计也表明, ASD 制剂体外溶出预测体内吸收仅有78%的准确率。了解体内吸收与体外溶出差异的本质原因, 有利于合理设计体外溶出条件, 并提高开发早期通过体外溶出快速判断体内吸收的效率。造成ASD 体内-体外相关性不理想的主要影响因素如下。
2.1 体内生理环境和体外溶出条件差异
ASD 制剂进入体内后, 其溶出与吸收会受到pH 环境改变和内源性表面活性剂的影响。现有药物中约75%为弱碱性药物[53], 对于弱碱性药物, 体内吸收过程中经历胃部和肠道的酸碱性环境变化, 可能导致溶解度骤降。而现有文献中的溶出条件往往是静态单一pH 条件, 无法模拟体内pH 的动态变化, 因此难以准确预测体内吸收。Indulkar 等[14]根据临床上药物推荐剂量、服药时推荐饮水量及体内吸收相关参数进行分析, 发现ASD 体内溶出时LLPS 现象是否发生, 受到药物的pKa、胃肠道pH 值及给药剂量等因素的综合影响。其次, 现有生物相关性溶出介质中, 包含胆盐牛磺胆酸钠和卵磷脂[54,55]。体内胆盐中占据大部分比例的并非牛磺胆酸钠, 内源性表面活性剂也并非只有牛磺胆酸钠和卵磷脂, 因此现有生物相关性溶出介质具有一定的局限性[54]。多篇相关工作表明, 不同胆盐对药物的增溶效果、抑制溶液析晶效果及对药物热力学活度和透膜的影响均不相同, 因此不同胆盐对药物的影响是相互不可代替的[41,54]。Enright 等[55]研究了其他内源性表面活性剂, 结果表明胆固醇抑制了阿扎那韦的成核, 而溶血卵磷比磷脂则促进了阿扎那韦的成核。
由于不同患者的生理环境有所不同, 体外溶出研究同样难以准确模拟。VanDen Abeele 等[56]通过对比单一服用Norvir®及Norvir®/质子泵抑制剂药物联用的血药浓度曲线, 模拟比较正常患者与胃酸分泌较少患者对ASD 制剂吸收的差异。结果表明服用质子泵抑制剂后, 5 位志愿者中有3 位药物吸收降低, 1 位药物吸收增加, 1 位吸收不变。该作者解释为质子泵抑制剂可能会减少胃肠液量, 从而改变了药物在胃肠道的吸收行为。
由于 ASD 体内吸收与体外溶出种种因素的不同, 现阶段ASD 制剂的体内-体外相关性并不能令人满意。理解ASD 体内外环境的差异, 有利于在开发早期通过体外溶出预测体内吸收, 从而促进ASD 制剂的开发。具体而言, 应在设计体外溶出条件时尽量使用与生理环境相关性较好的介质, 以便减少由于体内外差异所导致的相关性误差。
2.2 表观浓度与透膜通量之间的差异
现有文献中过饱和度的概念往往被定义为表观浓度与晶态溶解度之比, 但药物透膜通量并不一定与药物表观浓度直接相关, 而是与热力学活度直接相关。Raina 等[57]分别测定了添加/不添加维生素E 聚乙二醇琥珀酸酯 (D-α-tocopherol polyethyleneglycol 1000 succinate, TPGS) 的非洛地平混悬液的透膜通量, 结果表明虽然TPGS 的加入增加了药物表观浓度, 但两种混悬液中药物的热力学活度均与晶态药物相同, 因此透膜通量相同。由于透膜通量相较于体外溶出度更能反映ASD 体内吸收情况, 因此体外溶出实验中用表观浓度计算的溶出度存在一定的局限性[16]。
表观浓度与热力学活度的差异可能是由于ASD 在溶出过程中形成的LLPS和表面活性剂的增溶作用等。由于ASD 体内吸收时只有未电离的游离态药物才能被吸收, 而未电离的游离态药物浓度与热力学活度相关, 因此透膜通量与热力学活度而非表观浓度有关[29]。Lu 等[41]通过透膜实验证明了这一点, 当药物浓度低于无定形溶解度时, 透膜通量随药物浓度的增加而增加; 当浓度增加至无定形溶解度时, 体系产生LLPS 现象, 未电离的游离药物浓度保持不变, 透膜通量不再增加。同时Elkhabaz 等[17]的研究表明, 虽然泊沙康唑和阿扎那韦在6 种生理模拟介质中的表观浓度不同, 但最大透膜通量相似, 热力学活度相差不大。这说明溶液中游离药物的浓度是相似的, 表观浓度的改变源于形成的胶束中药物含量存在差异, 由于溶液中形成的胶束不能透膜吸收, 因此泊沙康唑和阿扎那韦在6 种生理模拟介质中的吸收透膜行为相似。同时辅料也可通过其他方式影响渗透, 如HPMC 可通过降低磷脂双层排列的紧密性促进姜黄素的膜渗透性[58]。
在 ASD 溶出时可能产生的LLPS 现象和内源性表面活性剂增溶作用的综合影响下, 药物表观浓度和热力学活度往往会解耦, 从而造成体外溶出表观浓度与体内吸收生物利用度不完全等效。此外, ASD 处方中可能存在促吸收的辅料也是造成体内外相关性差异的原因之一。因此评价ASD 时, 应当结合体外溶出实验中由表观浓度计算得到的溶出度和体外透膜吸收实验中测定的透膜通量, 两者相结合可提高体内外溶出与吸收行为的相关性。此外, Caco-2 细胞渗透性等相关实验有助于提高预测体内吸收的准确性。
2.3 体内吸收过程的复杂性
虽然体外溶出中常用非漏槽条件来模拟体内吸收时 ASD 形成的过饱和溶液,但仍与实际体内吸收环境有所差别。对于体外溶出, 溶液中的药物仅会由于溶液析晶而浓度降低; 但对于体内吸收, 药物浓度还受到吸收的影响。在一项他克莫司上市ASD 产品的对比研究中, 虽然体外溶出中Accord®结晶趋势高于Prograf®, 但在体内溶出时Accord®释放速率更快, 药物迅速被吸收, 降低了该ASD 制剂体内溶出过程中析晶的风险, 因此两种ASD 制剂的体内生物利用度相似[59]。
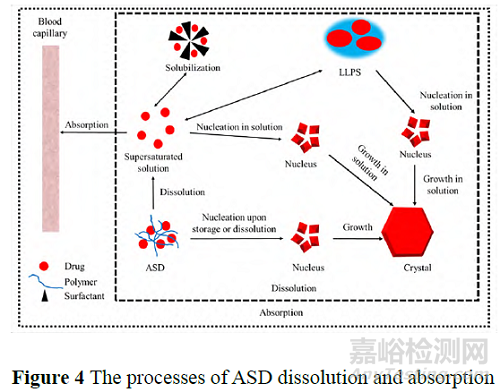
此外, 在ASD 体内吸收过程中, 可能在胃肠道中形成LLPS, 作为储库不断将药物释放至溶液中。Ueda 等[39]发现溶液中游离的布洛芬与LLPS 富药相中的布洛芬存在动态交换。这种动态的相互交换可能会影响ASD 的生物利用度。
Wilson 等[16]研究表明, 虽然恩杂鲁胺-PVP/VA (1∶1 w/w 和1∶9 w/w) ASD 和恩杂鲁胺-HPMCAS (1∶1 w/w) ASD 溶出形成了最大过饱和度相似的溶液, 但仅恩杂鲁胺-PVP/VA (1∶9 w/w) ASD 溶出时形成了LLPS, LLPS 的储库效应使得药物在溶液中浓度在较长时间维持较高水平, 生物利用度显著提高。
相较于体外溶出, 体内吸收过程中药物的行为更复杂, 使得预测生物利用度更困难。因此在现有溶出装置中, 可考虑增加吸收室或运用两相溶出评价ASD的体外释放与吸收行为。体外溶出实验尽可能模拟体内吸收过程, 建立体外溶出-吸收过程的实验方法。
2.4 其他因素
虽然体外溶出相关条件可精确控制, 但服药方式的不同仍可能导致ASD 体内吸收不同。Pas 等[60]研究了研磨对上市ASD 制剂体外溶出的影响, 结果表明研磨使得ASD 体外溶出改变, 但对体内吸收的影响仍需进行生物等效性研究。Sironi 等[61]发现ASD 制剂与水共同给药于大鼠时, 生物利用度与晶态相同; 而如果仅将ASD 制剂给药于大鼠, 生物利用度相较晶态增加了4 倍。该作者推测,ASD 制剂的体内吸收行为受到以下几点因素的协同影响:制剂溶出过程中发生结晶、溶出过快使得药物在胃中析出,肠道中供吸收的药物浓度降低以及胶囊壳及其内容物被溶解与分散, 阻碍了与肠道黏膜更密切的相互作用。其次, 非线性药动学也是造成体内外不相关的因素之一。市售ASD 制剂依曲韦林(intelence) 存在非线性药物动力学特征, Litou 等[62]发现相较100 mg 规格制剂,200 mg 规格制剂血药浓度被低估。此外, Liu 等[63]认为溶出介质体积和实际胃肠道液体积的差异以及流体动力学上的差异也可能影响ASD 的体内外相关性。体外溶出的溶出介质体积往往较大且充分搅拌, 因此ASD 片剂崩解较快, 药物迅速释放。然而在体内溶出和吸收过程中, 由于溶出介质体积较小, ASD 片剂可能无法足够快地崩解和分散, 从而导致体内外的差异。
3 结论与展望
ASD 可显著提高难溶性药物的溶解度和溶出速率, 但ASD 溶出时可能发生表面结晶、去过饱和与LLPS 等现象, 导致丧失溶出优势或使得溶出行为复杂化,难以预测。因此在ASD 开发过程中, 即使ASD 的体外溶出达到预期的释放速率并维持较高的过饱和状态, 仍需充分考虑ASD 溶出和吸收过程中的各类影响因素和复杂性,以便及时调整和优化处方和工艺。此外, 核磁共振、宽频介电谱、原位光谱和成像等分析技术的发展有助于制剂开发工作者更好的理解ASD 在溶出和吸收过程中的动态演变和内在机制,从而更有效的进行产品过程分析和质量控制。
其次, 由于体内外环境的差异、过饱和度与透膜通量之间的差异、体内吸收过程的复杂性及服药方式等因素的综合影响, ASD 体外溶出往往并不能完全准确预测体内吸收情况。因此在实际开发过程中, 应注重体外溶出与体外透膜吸收等其他实验相结合, 同时对溶出介质、溶出装置等研究方式加以改进, 以便更真实地模拟体内吸收过程, 有利于提高预测ASD 体内吸收的成功率。
此外, 计算机分子模拟和大数据机器学习等方法也有利于ASD 制剂体内外相关性预测。越来越多的案例和研究报道通过药物和载体的结构、溶出数据等已有数据, 进行机器学习, 建立预测数学模型, 预测ASD 的溶出情况[64]。
随着对 ASD 溶出和吸收过程的深入研究, 以及相关表征手段和计算机信息技术的发展, 有望更科学地设计ASD 的处方及工艺,从而避免ASD 出现结晶,更有效地增加药物吸收, 提高体内外相关性预测的准确性。相信未来会有更多ASD 产品上市, 进一步提高难溶性药物的成药性,服务患者并造福社会。

来源:Internet


