您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-04-20 06:30
对于色谱工作者而言,面对复杂成分的检测,分离是首要的关注点。无法有效分离意味着无法进行准确的定性、定量检测。
说起分离,有必要再介绍一下色谱。了解、理解并掌握色谱分离原理很重要,对于大多数接触了色谱分离的人而言,应该了解的基于不同分离原理的主要色谱技术包括吸附色谱、分配色谱、离子交换色谱、亲和色谱、大孔吸附树脂、凝胶色谱等。如果说上面这一系列名词还稍显陌生,那么根据分离方法所分类的纸色谱法、薄层色谱法、柱色谱法、气相色谱法、高效液相色谱法等,应该更广为熟知了。
无论是何种分离色谱,对于目标成分的分离均有相应要求。以中国药典通则0500色谱法项下的相关描述为例,薄层色谱中要求“除另有规定外,分离度应大于1.0”,高效液相色谱法、离子色谱法及气相色谱法等要求“除另有规定外,待测物质色谱峰与相邻色谱峰之间的分离度应不小于1.5”,分子排阻色谱法中要求“除另有规定外,分离度应大于2.0”。
从上述要求中可以直观的看出,不同色谱技术对于分离度的要求不尽相同,至于个中缘由我们暂不揣度。只是有一点很重要:分离度不是指导原则等文件规定死的一个参数,正如大多技术参数一样,始终是为了满足使用目的而进行的“私人订制”,量后裁!
本文主要以高效液相色谱法为例,抛砖引玉谈一下系统适用性中“分离度”这个参数,以期对该参数有一个更为深入的思考和认知,其中的案例也希望能给实际工作一些借鉴。
一、什么是基线分离?
基线分离是每一个色谱分离工作者在兼顾效率前提下的终极追求。理论上,当分离度等于1.5时,即可认为达到了基线分离。为什么是“即可认为”,而不是绝对认为呢?这里面涉及到正态分布/高斯分布,3σ,6σ等概念。这些概念稍显顽固,我们给予记过并留待察看,暂不深入追究。用点色谱参数里常用的名词,再加一点通俗语言来说就是,对称因子为1.0的两个高矮胖瘦一模一样的色谱峰,当分离度为1.5时可认为是基线分离。比如,下图1显示的某药典给出的有点标准的示例图。

图1 基线分离示例
但是,理论指导实践,实践是检验真理的唯一标准。比如说,上面对于基线分离的描述情形,在实际工作中基本不可能,而且说“基本”是为了满足“一切皆有可能”的严谨,否则可以直接说“根本不可能”。因为德国哲学家莱布尼茨说,世界上没有完全相同的两片树叶,而每一个色谱工作者都可以说,世上没有完全相同的两个色谱峰。
实际情况是,往往相邻色谱峰各有千秋,导致同一个分离度会出现完全不同的分离效果。比如下图2,两个不同比例的色谱峰,在不同分离度情况下的分离表现,在分离度0.6时尤为明显:当后峰是前峰的1/16时,还能看出是两个峰吗?
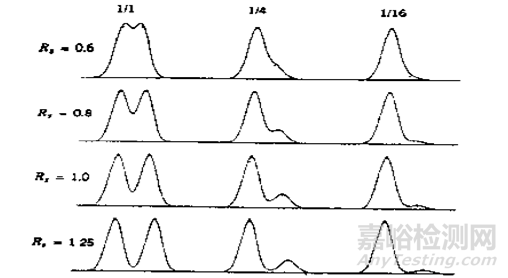
图2 分离度及谱峰相对大小(面积)与分离度关系《实用高效液相色谱法的建立》
既然如此,那我们就不要再执念于分离度1.5了,也不要自欺欺人的出具一个显示分离度大于1.5的色谱报告而不看实际分离效果了。
二、分离度如何定?
很多人都做过仿制药吧,或者改良型新药,甚至创新药!99.9%的核心技术思想离不开模仿、借鉴,还有0.1%留给那些从0到1的“传说”。同样的,我们这个问题,不妨看看别人家的孩子是如何优秀解决的。
下表1列举了从中国药典以及欧洲药典中随机摘录了一些典型的实例,对于分离要求可以说是“五花八门”。当然,这里对于具体品种为何制定了当前的分离要求,以及做了哪些研究,我们暂不做深究,也确实有心无力,无从下手,仅仅从表象来看个热闹。
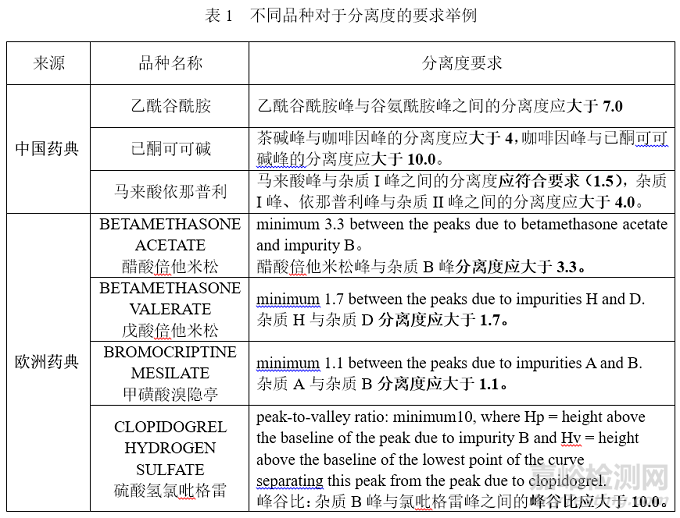
如果说中国药典中的例子还算中规中矩的拓宽了我们对分离度1.5的认识,那欧洲药典分离度3.3、1.7、1.1是什么操作?这种有零儿有整儿的分离度是怎么搞出来的儿?峰谷比又是什么操作,跟分离度有什么区别,我该怎么选?看完以后什么感觉?眼花缭乱,再加上点眩晕,有点上头!
带着一脑门儿的问号,总感觉,法典里透着点草率,严谨中不失那么点随意。要不是看在欧洲药典的面子上,领导不得骂的你找不着北?
既然欧洲药典都可以这么干,那我们能不能也模仿、借鉴一下?能!
而且,在这个彰显个性,鼓励创新的时代里,建议不要做分离度1.1、3.3这种纯仿制,也不要假模假式的做一些分离度1.2、3.4这种me-too,可以尝试做一些分离度2.2、4.4这种me-better,或者大胆一点做一些分离度1.03、2.21这种四不像。
三、如何证明合理性?
实际工作中也接触到不少人有这个想法:当分离度连中国药典那除另有规定外的1.5都达不到时,开始动了定分离度1.0、1.2的心思。但往往只是大笔一挥而已!比如,方法开发及方法验证时实际分离度为1.4,为了日后方法系统适用性通过性的保险起见,怀着一丝的忐忑,带着欧洲药典的一丝安慰,标准制定时写下了1.2,大胆一点的直接来个1.0。
合理吗?有人说之所以定了1.2,是来源于耐用性验证时在某条件下的最差分离度。暂且不去推测该耐用性试验是如何设计的,但我相信大多数情况下对于制定1.2分离度的证明性是不够的。
当然,不仅是达不到分离度1.5时的退让,更有很多达到1.5后的前进,比如分离度应大于10.0。这些数值的制定不仅仅是一个耐用性的探索,更不是冒险或保守之类的估计,而是需要具体的试验设计去验证。
以下提供一个参考实施例,用于说明制定分离度时的考虑。
以“甲磺酸溴隐亭”有关物质分析方法为例,色谱条件如下:
色谱柱:Octadecylsilysilica gel for chromatography R,4mm×0.12m,5μm
流动相A:0.791g/L的碳酸铵溶液
动相B:乙腈
梯度洗脱表:
|
时间(min) |
流动相A |
流动相B |
|
0-30 |
90→40 |
10→60 |
|
30-45 |
40 |
60 |
流速:2ml/min
检测波长:300nm
进样体积:20μL
系统适用性要求(图3):杂质A与杂质B分离度应大于1.1。
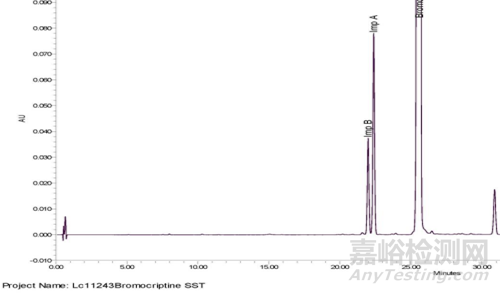
图3 “甲磺酸溴隐亭”有关物质分析方法系统适用性图谱
根据以上信息,我们可以推断的是,实际分离效果肯定不止1.1,这只是一个最低可接受限度。因此在正常色谱条件下对方法进行完整验证后,针对希望拟定的分离度再次进行试验设计以验证合理性。
方法学验证设计(针对分离度或峰谷比制定)简述如下(如有遗漏谬误还请自行脑补修正,朝着目标前进):
1、拟定分析方法耐用性的关键参数:碳酸铵溶液浓度、流速、柱温、色谱柱及仪器。
2、拟定关键耐用性参数在日常检测中可能出现的偏离,比如碳酸铵溶液浓度由于配制过程导致的误差,由于仪器本身性能导致的流速及柱温控制差异,色谱柱批次间的差异,不同仪器(型号、品牌)之间的差异。这些差异均可以根据经验及相关的性能确认(PQ)进行预先估计,在估计的基础上适当放宽考察。
3、在上述各种拟定的偏离条件下进行分离度考察,确定最差分离度,比如说是在流速2.2ml/min时分离度为最差的1.1。
4、在流速2.2ml/min,分离度为1.1的条件下,设计杂质A与杂质B的灵敏度(检测限、定量限),线性与范围,准确度(加标回收)试验。此时,我们甚至可以在该条件下兼顾其他杂质的再次验证。
5、在最差分离度条件下,各项验证均满足拟定的验证要求时(不一定非得完全按照中国药典9101的要求),可以合理的制定所希望的分离度或峰谷比。
此外,尚需说明一点的是,耐用性不是通过性验证,无需拟定范围后追求耐用性试验的通过。耐用性的参数应在方法开发阶段已完成边界的探索,并进行了初步的验证;在方法学验证阶段根据开发时的边界,适当调整符合实际使用场景的参数偏离范围并进一步的确认。
因此,需要根据具体情况,在参数调整后设计不同程度的耐用性验证试验,并没有一个SOP似的参考模板,这是该项验证与其他验证项目最大的不同,也是我们制定非常规要求分离度(比如分离度1.0,10.0,峰谷比5.0等)的基础。
四、结语
上述的实施例仅仅是以偏概全,抛砖引玉,是否有实际意义以及参考性不敢言定,但若能引发对分离度的一些新的认识和思考已是万幸。

来源:Internet


