您当前的位置:检测资讯 > 行业研究
嘉峪检测网 2022-05-29 00:05
使用真实世界证据支持全球医疗器械监管决策现状
Use of Real-World Evidence to Support Global Regulatory Decision-Making for Medical Devices
摘 要 / Abstract
医疗器械的监管审批传统上主要依赖于被称为临床研究金标准的随机对照试验(RCT)。快速发展的真实世界数据(RWD)和真实世界证据(RWE)领域在以数据为驱动的创新方面为监管机构和医疗行业带来了巨大的希望。在适当的条件下,充分利用高质量的RWD,作为RCT数据的补充,可以实现更高效地医疗器械开发,并优化产品全生命周期的监管决策。然而,在全世界范围内,监管部门对于RWD/RWE的可接受性和适用性仍然存在差异。本文回顾了RWD和RWE的现有全球指南,并总结了美国系统性地使用RWE支持医疗器械监管决策的宏观政策要素和相关考虑因素。
Traditionally, regulatory approval for medical products has predominately relied on randomized controlled trial(RCT), the gold standard for clinical research. The fast-evolving field of real-world data(RWD) and real-world evidence(RWE) holds great promise in data-driven innovation for both regulatory authorities and industry. Under the right conditions, unleashing the power of high-quality RWD, as a complement to RCT-derived data, enables more efficient medical device development and streamlines regulatory decision-making across the product life cycle. However, there remains a gap in the acceptability and applicability of RWD/RWE for regulatory use worldwide. The current report reviews the existing global guidance on RWD and RWE and summarizes the principal elements and considerations associated with the systematic use of RWE in support of regulatory decision-making for medical devices in the United States(U.S.).Traditionally, regulatory approval for medical products has predominately relied on randomized controlled trial(RCT), the gold standard for clinical research. The fast-evolving field of real-world data(RWD) and real-world evidence(RWE) holds great promise in data-driven innovation for both regulatory authorities and industry. Under the right conditions, unleashing the power of high-quality RWD, as a complement to RCT-derived data, enables more efficient medical device development and streamlines regulatory decision-making across the product life cycle. However, there remains a gap in the acceptability and applicability of RWD/RWE for regulatory use worldwide. The current report reviews the existing global guidance on RWD and RWE and summarizes the principal elements and considerations associated with the systematic use of RWE in support of regulatory decision-making for medical devices in the United States(U.S.).
关 键 词 / Key words
真实世界证据;医疗器械监管决策;创新性监管审批
real-world evidence; medical device regulatory decision-making; innovative regulatory approval
真实世界数据(real-world data,RWD)是指来源于日常所收集的各种与患者健康状况和(或)诊疗及保健有关的数据,包括电子健康记录(electronic health record,EHR)、医保支付数据、疾病登记数据和患者生成的数据等[1-2]。真实世界证据(real-world evidence,RWE)是指通过对适用的RWD进行恰当和充分的分析所获得的关于药物的使用情况和潜在获益-风险的临床证据[1-2]。随着医疗数据数量的迅速增长和医疗信息技术的快速发展,RWD/RWE已成为推动全球医疗创新不可或缺的部分,并能够为医疗器械性能和器械使用相关的临床结果提供新的参考[3]。传统上,医疗器械的治疗疗效评价主要依靠于随机对照试验(randomized controlled trial,RCT),这是高质量临床研究的金标准。RWD常被用于上市后的不良事件监测以及辅助器械长期安全性的监管决策。然而,近年来,随着RWD受到越来越多的关注,监管决策者和医疗器械开发者都意识到了RWD/RWE在医疗器械全生命周期当中的重要性,尤其是在有效性评估方面[2,4]。在适当的条件下,利用高质量的RWD,作为RCT结果的补充,能够促进医疗器械创新,加速患者获得临床急需的器械治疗,并优化监管决策过程[2,4-5]。
与传统的RCT相比,RWE有其独特的优势。RCT的受试者筛选受到严格的纳入和排除标准的限制,可能导致较低的外部有效性。而设计良好的RWE能够为监管决策者提供在真实临床实践中更广更多样人群中器械的安全性和有效性的补充信息[6]。此外,RWE可填补RCT未能解决监管问题中的几个空白。例如,通过更长的随访期发现额外的治疗效果或风险,调查患者特征、成本效益和依从性模式,并揭示传统临床试验中未能满足的医疗需求[7-8]。但是,RWD存在多数据源的协调不充分、互操作性不足、研究能力和需求差异较大、缺乏系统性的数据质量评估工具等问题,为RWE的进一步发展带来了巨大的挑战[9-10]。因此,需要所有利益相关者之间进行广泛合作,包括监管机构、药械厂商、医疗行业和患者等,以对现有RWE框架达成共识,并制定相关的系统化和标准化的方法。
由于认识到RWD/RWE的潜在价值,世界各地的监管机构也相继发布了相关指南,并对利用RWE进行监管申请的医疗器械给予了审批。然而,目前还没有针对RWD/RWE的可接受性和适用性的国际公认指南。本文全面地回顾了与RWD/RWE相关的全球指南。结合RWE支持医疗器械监管决策的实例,总结美国医疗器械监管审批的相关核心要素。
01方 法
本文在Pub Med、MEDLINE和Embase电子数据库中对RWD和RWE的现有文献进行了检索。搜索关键词为“real world evidence”“real-world data”“regulatory approval”和“r e g u l a t o r y d e c i s i o n making”。纳入的研究仅限于用英文或中文发表,且发表年份为2005年及以后。在以下国际监管机构网站中对RWD/RWE相关监管指南及法规进行检索,包括国家卫生健康委员会(National Health Commission of the People's Republic of China,NHC)、国家药品监督管理局(National Medical Products Administration,NMPA)、美国食品药品监督管理局(Food and Drug Administration,FDA)、欧洲药品管理局(European Medicines Agency,EMA)、药品局总部(Heads of Medicines Agencies,HMA)、加拿大卫生部(Health Canada)、加拿大药品和卫生技术局(Canadian Agency for Drugs and Technologies in Health,CADTH)、日本药品及医疗器械综合机构(Pharmaceuticals and Medical Devices Agency,PMDA)。
02结 果
2.1 RWE全球指南发展概况及展望
2.1.1 美国
美国是大力鼓励将RWD/RWE纳入监管审批程序的先行者之一。作为《2009美国复苏与再投资法案》(American Recovery and Reinvestment Act of 2009)的一部分,《卫生信息技术促进经济和临床健康法案》(Health Information Technology for Economic and Clinical Health Act),旨在创建可互操作的医疗系统并提供可访问的患者健康信息,推动了EHR的实施[11]。根据美国国家卫生统计中心(National Center for Health Statistics)统计,截至2017年,已有86%的美国临床医生在使用EHR系统[12]。EHR的高使用率进一步推动了RWD数量的大幅增加。在2016年《21世纪治愈法案》(21st Century Cures Act)颁布之前,RWE常被FDA用作于上市后的长期安全性评估。于2008年启动的哨点计划(Sentinel Initiative),通过系统性地收集RWD(包括EHR、疾病登记数据和医疗索赔数据等),建立了国家风险识别和分析系统,以主动监测医疗器械和药品的上市后安全性[13-14]。随后,FDA于2013年发布了《使用电子医疗数据集设计和报告药物流行病学安全性研究的最佳实践》(Best Practices for Conducting and Reporting Pharmacoepidemiologic Safety Studies Using Electronic Healthcare Data Sets)[15]。哨点计划为RWE纳入监管决策提供了重要的前期基础。2016年《21世纪治愈法案》的颁布被认为是RWE的一个重大转折点。为了促进医疗产品创新并倡导在监管决策过程中扩大RWE的使用,《21世纪治愈法案》从法律层面要求并指导FDA实施RWE使用评估方案,并要求发布RWE用于安全性和有效性评估的相关指南[16]。为响应《21世纪治愈法案》,FDA于2017年发布了《使用真实世界证据支持医疗器械监管决策指南》(Use of Real-World Evidence to Support Regulatory Decision Making for Medical Devices),系统性地说明了RWE在支持医疗器械监管决策方面的可接受性和适用性[17]。随后,《FDA真实世界证据计划框架》(Framework for FDA’s Real-World Evidence Program)和《在临床研究中使用电子健康记录的行业指南》(Use of Electronic Health Records in Clinical Investigations Guidance for Industry)于2018年发布[1,18]。除了医疗器械,FDA还将RWE辅助监管决策的积极倡导扩展到了药物和生物制剂领域,并于2019年发布了《使用真实世界数据和真实世界证据向FDA递交药物和生物制剂指南》(Submitting Documents Utilizing Real-World Data and Real-World Evidence to FDA for Drugs and Biologics)[19]。此外,根据《处方药使用者付费法案》(Prescription Drug User Fee Act),FDA预计在2021年10月前公布一份指南草案,阐明如何使用RWE作为监管决策中评估药物有效性和安全性的重要工具[20]。由于FDA的大力推广,根据美国器械与放射卫生中心(Center for Devices and Radiological Health)的报告,2017年RWE用于上市前和上市后的监管决策数量比2015财年基准增加了193%[21]。对于医疗行业来说,一项由德勤公司在美国进行的调查显示,医疗器械公司已计划优先考虑将RWE纳入产品研发过程[22],且对RWE重要性的认知可部分归因于FDA颁布的指南和声明[23]。
2.1.2 欧洲
EMA是全球大力推动RWE用于监管决策的另一个主要机构,并于2013年启动了GetReal项目。GetReal积极地与RWE的各方利益相关者合作,如医疗决策者、制药公司和学术研究者,创建了收集RWD的前沿工具和资源,并提出了RWE在促进监管决策方面的可接受性的考虑[24]。2014年,EMA实施了适应性审评(adaptive pathway)的试点项目,将RWE作为一种协助有条件上市许可的工具,以加速患者的治疗过程并解决未满足的医疗需求[25]。该方法强调了真实医疗环境中的有条件批准上市机制,以积累RWD作为临床试验数据的补充,并促进监管决策。2017年,EMA和HMA成立了联合大数据工作组(big data taskforce),旨在加强RWE在产品全生命周期内为监管决策提供有效信息的能力[26]。作为该工作组总结报告的一部分,EMA和HMA于2019年联合发布了《观察性数据(真实世界数据)亚组报告》[Observational Data (Real World Data) Subgroup Report],从监管的角度描述了倡导应用RWD的挑战和建议[27]。尽管EMA和HMA在将RWE纳入监管决策的方面做出了上述努力,但迄今为止尚未发布任何相关指南,且欧洲各地的国家和机构对于RWE在监管方面的应用也持不同态度[28-29]。一项调查欧洲6家卫生技术评估(Health Technology Assessment)机构使用RWD政策的研究表明,各机构对于RWE在相对有效性评估中的应用存在明显差异[28]。2020年,EMA发布了《2025监管科学战略》(Regulatory Science Strategy to 2025),把在监管决策中系统地使用高质量RWD作为一项战略目标,并强调了与各利益相关者共同制定RWE/RWD相关指南的必要性[30]。
2.1.3 中国
近年来,我国通过多维度创新,致力于RWE在监管审批流程中的应用。2017年,中共中央办公厅、国务院办公厅发布《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,以通过适应性和创新性的方法加速医疗器械和药品的评估和批准工作[31]。作为响应,国家药品监督管理局药品审评中心(Center for Drug Evaluation of NMPA,CDE)于2019年发布了《真实世界证据支持药物研发的基本考虑(征求意见稿)》[32]。2020年,NMPA进一步发布了《真实世界数据用于医疗器械临床评价技术指导原则(试行)》,明确了相关定义,并描述了用于医疗器械开发的监管审批过程中产生RWE的RWD质量要求和研究设计[33]。在这两个指导原则的基础上,由四部门联合发布的《关于支持建设博鳌乐城国际医疗旅游先行区的实施方案》强调了RWD的重要性,并为RWD向RWE转化提供了一个开拓性的平台,以支持进口医疗产品的监管批准[34]。随着RWD试点项目的实施,国外已上市的临床急需进口医疗器械和药品无需在国内进行额外注册审批就可供患者使用,并产生了可用于监管决策的临床RWE。2020年和2021年,NMPA和CDE分别发布了《真实世界证据支持药物研发与审评的指导原则(试行)》和《用于产生真实世界证据的真实世界数据指导原则(试行)》,进一步阐明了RWE用于评估监管决策的基本原则,并概述了用于产生RWE的高质量RWD的使用原则和范围[2,35]。
2.1.4 其他国家
在医疗器械研发过程中使用RWE的潜在价值也逐渐被世界各地的其他监管机构所认可。加拿大卫生部联合主要利益相关者共同制定了RWE策略,并确定了在产品全生命周期中使用RWE的机会。具体地,加拿大卫生部提出《优化加拿大医疗器械生命周期中真实世界证据使用的策略》(A Strategy to Optimize the Use of Real-World Evidence Across the Medical Device Life Cycle in Canada),旨在通过优化高质量RWE的使用,提高医疗器械在产品全生命周期中的可及性、可负担性和使用的适当性[36-37],并计划于2019年开始起草与医疗器械评估相关的RWE指南。同样的,PMDA已采取重要措施,逐渐转移RWE在监管决策中的使用重心。在日本,RWE在监管批准方面的应用以往主要基于逐案处理的方式,并以批准后的安全性评估为重点。为了推动扩展药械监管审批纳入RWD的使用,PMDA于2021年初发布了《疾病登记数据用于监管提交的基本原则》(Basic Principle for Utilization of Registry Data for Regulatory Submission)和《确保数据可靠性的考虑要点》(Points to Consider for Ensuring the Data Reliability),并制定了RWD工作组提供RWD/RWE相关建议,以鼓励药械产品开发者在产品全生命周期的监管申请中使用RWD[38]。
2.2 医疗器械监管批准的宏观标准及要素
尽管受到了全球性的关注,但RWE在支持监管决策方面的系统性应用仍处于起步阶段,特别是在医疗器械方面。FDA和EMA作为全球2个主要的监管机构,积极参与优化了RWE用于医疗产品全生命周期的监管决策。然而,欧洲主要应用RWE用于加快罕见病用药的审批流程[39-40]。在世界其他国家,RWE辅助医疗器械监管决策的可接受性主要以逐案制度审批。FDA迈出了重要的一步,发布了RWD/RWE相关指南,系统性地阐明了用于评估RWD是否具有足够质量以产生有效的RWE来支持医疗器械的安全性和有效性的核心原则和考虑要素。在《使用真实世界证据支持医疗器械监管决策指南》中[17],FDA指出了使用临床RWD成功获批医疗器械的关键要素,即RWD和RWD数据源的相关性和可靠性。
2.2.1 相关性
在评估RWD和RWD数据源的相关性时,FDA强调了选择适当的RWD数据源的重要性,这是影响对数据质量总体理解的要素之一。常见的RWD数据源包括但不限于:纸质健康档案或EHR、医保数据库和特定疾病或治疗数据库(如疾病登记处)。用于评估器械性能的充分且适当的RWD数据源应能回答相关监管问题或满足监管要求。由于RWD通常是为了非监管目的而开发,在现有的RWD数据源中,FDA将进一步确定单个数据元素及其范围,以判断是否足够用于监管审批。具体而言,FDA将对以下关键因素进行相关性评估:(1) RWD能否在适当的人群中充分捕捉关键信息细节,包括设备的使用信息、暴露和相关的临床结果等;(2) RWD数据元素能否结合有效且适当的分析方法回答特定监管问题;(3)使用临床/科学的判断能否解读RWD和RWE。其中,可解读性的重要考虑因素包括但不限于:RWD数据源对相关目标人群的代表性和普遍性;RWD数据源的范围(地区性、国家性或国际性);RWD数据源能否采集设备标识信息;能否采集患者对于设备的使用情况;是否具有充足的RWD数据元素以评估相关临床结果和调整混杂因素;RWD研究设计、方案和(或)分析计划是否适合解决相关监管问题;RWD是否具有充分采集患者病史、既往情况和随访信息的能力(RWD的充分连续性)等。
2.2.2 可靠性
RWD的可靠性主要取决于数据累积和数据保证(质量控制)。RWD数据源应具有经验证的和标准化的数据收集、汇总、管理和记录方法,包括操作手册、数据字典、通用病例报告表等。评估数据累积/收集的要素包括但不限于:RWD数据源是否具备完整且准确地收集RWD的能力(合格的人员、明确的流程、工作培训和支持等);是否使用通用数据采集表格和通用定义框架(数据字典);是否遵守关键数据收集的共同时间框架;患者筛选和登记标准是否具有普遍性和代表性;用于采集数据元素的来源和技术方法(与EHR或保险索赔数据库的链接方法等);是否使用数据质量审计程序等。此外,数据保证对于RWE的证据质量至关重要。为保证RWD的质量,使用系统化且标准化的方法收集和清洗RWD不可或缺。FDA将考虑以下数据质量关键因素:数据完整性;数据一致性(跨站点和随时间变化);与可验证的数据源相比,采集的数据元素的准确性;是否遵守数据源的验证程序以及数据采集和记录程序;是否具有数据监控措施的评估等。在这些因素中,数据的准确性尤为重要。尽管数据清理和交叉引用等常用方法能够确认一致性并评估数据完整性,但无法完全保证数据的准确性和真实性。为进一步确保RWD质量,FDA建议使用各种数据验证程序,包括数据监测和数据质量审计计划。
总体来说,被FDA所认可的用于医疗器械监管决定的RWD/RWE,通常是适用于监管目的的[41]。FDA根据相关性和可靠性(数据累积和数据保证)的原则对RWD数据源进行评估,并在遵守上述标准方面具有很好的灵活性。因此,FDA强调了在利用RWD进行研究和申报前,使用预提交流程及充分沟通的重要性。此外,用于评估RWE支持医疗器械监管的标准还应建立在当前FDA已有的证据标准基础上,包括公认的数据标准以及RWD数据分析和评估临床及统计意义的合理方法。由于现有的RWD数据源容易产生潜在的偏倚,并限制所产生RWE的强度,应采用科学稳健的研究设计方法,以及与传统临床试验内容和标准相同的、预先制定的研究方案和分析计划。
2.3 RWE在医疗器械监管批准中的应用示例
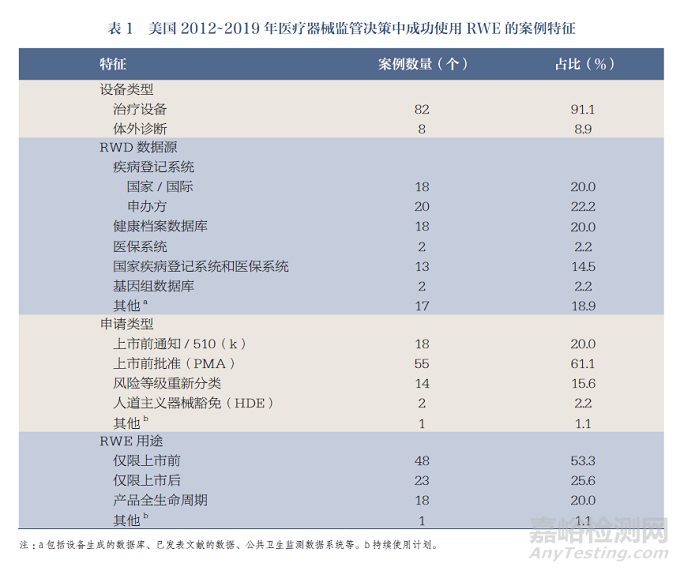
2021年3月,FDA发布了90个2012~2019年期间RWE成功应用于医疗器械监管决策的案例。这90个案例中所使用的RWD都对上述相关性和可靠性的标准给予考虑[42]。其中,82个(91.1%)为治疗设备,适应症包含心血管疾病、青少年特发性脊柱侧弯和近视等。值得注意的是,其余8个(8.9%)使用RWE对创新性的体外诊断设备进行了评估,包括下一代测序技术、荧光素酶活性测定和个人基因组测试。在RWD数据源方面,38个(42.2%)采用了国家、国际或申办方的疾病登记系统,18个(20.0%)使用了病历数据库[EHR、电子病历(electronic medical record,EMR)或病例回顾],13个(14.4%)结合了国家疾病登记系统和医保系统,17个(18.9%)采用了其他RWD数据源,如来自美国以外的设备生成的数据和已发表文献的数据。此外,超过一半(61.1%)的申请类型为上市前批准(premarket approval,PMA)申请,20.0%使用了RWE支持产品全生命周期的监管批准(表1)。
2.3.1 RWD数据源作为支持性平台开展RCT辅助监管决策的示例
Volcano血管内超声诊断仪,于2014年获批用于定性和定量评估冠状动脉和外周血管的血管形态。该仪器可连接相应的压力导丝,结合血流储备分数(fractional flow reserve,FFR)和瞬时无波形比率(instantaneous wave-free ratio,iFR)的混合方法作为冠状动脉功能学评价指标,以辅助血管临床决策。2017年,申办方向FDA提交了510(k)上市前通知申请,旨在通过一项美国境外的、嵌入疾病注册登记系统的、多中心、随机、对照、开放标签的临床试验(iFR-SWEDEHEART试验),将混合评价指标替换为i FR的二分法切点指标。瑞典冠状动脉造影和血管成形术注册登记系统(SCAAR)是瑞典根据推荐疗法评估的心脏病循证护理增强和发展网络系统(SWEDEHEART)的一部分。作为一个全国性的、基于网络的、综合的疾病注册登记系统,SCAAR从瑞典全部31个冠心病医疗中心和冰岛的1个中心,前瞻性地收集有关急性冠状动脉综合征标准治疗的临床数据,包括患者人口统计、病史、干预措施、血管造影结果和随访信息等[43]。为了确保数据质量,SCAAR系统采用了指南手册、人员培训、包含标准化定义的数据字典以及技术咨询等措施,还实施了包括错误检查、与国家SWEDEHEART系统交叉验证随机选择的变量的数据监测活动、强制性数据输入等策略,以确保数据的准确性和完整性[43-44]。
iFR-SWEDEHEART RCT嵌入用于数据收集、随机化和随访的SCAAR系统中,并根据预先指定的纳入和排除标准,在该系统内进行受试者招募[44]。该试验基于非劣效性设计,从SCAAR系统中选出2017名受试者,并按照1∶1的比例,利用SCAAR网络平台将符合条件的受试者随机分配至接受iF R或FFR指导的血管重建术组中。主要有效性终点为已在SWEDEHEART注册登记中收集的,由手术后12个月内全因死亡率、非致命性心肌梗死或非计划的血管重建术组成的复合发生率。基线人口统计数据和手术相关的临床信息直接从SCAAR中获得。此外,未包含在SCAAR中的i FR-SWEDEHEART试验特定数据将通过嵌入SWEDEHEART系统的在线调查问卷中被单独收集。研究结果显示了12个月时i FR组和FFR组主要有效性终点的非劣效性,并发现了i FR组不良反应显著减少[45]。这项嵌入在SCAAR注册登记系统中的i FR-SWEDEHEART临床试验的结果支持了FDA对实质等效性的确定,并辅助了该设备成功修改适应症使用声明,以使用i FR血管内压力指数指导血管重建手术[45]。该设备的申请批准也是RWD数据源作为支持性平台开展RCT辅助监管决策的示例。
2.3.2 RWE在评价医疗器械扩大儿科人群适应症修改的示例
在过去,由于伦理和法律方面的考虑,被视为弱势群体的儿科人群一般难以被纳入传统临床试验中进行有效性和安全性的研究,特别是罕见病[42]。RWE能够在真实世界的临床实践中就医疗器械在儿童中的使用情况提供实用且有价值的见解,以辅助包括适应症扩展和标签修改在内的监管决策。以下示例展示了RWE在儿科人群中,评价心脏辅助装置产品适应症修改的关键性证据。
EXCOR儿科心室辅助装置(EXCOR)最初于2011年通过了人道主义器械豁免(humanitarian device exemption,HDE)申请,旨在为患有严重孤立性左心室或双心室功能障碍的儿童患者提供心脏机械性循环支持,作为心脏移植前的过渡[46]。HDE是针对罕见病医疗器械上市审批的一种途径。根据《21世纪治愈法案》,在美国每年用于诊断或治疗不超过8000人疾病的医疗器械被定义为人道主义使用器械(humanitarian use device,HUD)[16]。对于通过HDE途径的上市申请,由于罕见病在人群中的特殊性,FDA主要评价HUD的安全性和可能的获益,并免除对HUD有效性证明的规定,以加速上市批准[47]。在EXCOR的原始HDE申请中,申办方通过一个前瞻性、多中心、单臂临床试验[研究性器械豁免(investigational device exemption,IDE)试验],结合体外生命支持组织(extracorporeal life support organization,ELSO)系统中获取的RWD作为体外膜肺氧合(extracorporeal membrane oxygenation, ECMO)历史对照组,说明了该装置的安全性和可能的获益[46]。ELSO是收集北美接受ECMO治疗患者信息最全面的国家注册登记系统,致力于通过临床数据收集提高患者护理质量[48-50]。ELSO系统性地收集了包括儿童和成人患者的人口统计信息、既往病史、诊断和治疗、使用体外生命支持设备的适应症等信息[49]。为了提高数据收集的有效性、价值和效率,成立了ELSO注册登记小组委员会[50]。使用标准化的数据收集表格以及数据库定义手册以达到各个参与中心数据收集的一致性,提供人员培训,并要求强制性的数据库录入考试以保证数据收集的可靠性[49-50]。数据准确性是通过对随机选择10%的患者数据进行外部验证来确保的[50]。此外,ELSO还实施了基于网络平台的自动化数据完整性监控计划,以简化数据收集流程并最大限度地减少缺失数据[48,50]。IDE单臂临床试验共包括48名接受EXCOR治疗的受试者,其年龄在30天到16岁之间。根据预先指定的6个变量,包括年龄、体重、初步诊断、呼吸机状态、正性肌力药物使用和先前心脏骤停,研究对从ELSO注册登记系统中选择的、作为心肺衰竭标准治疗的ECMO历史对照组进行了倾向评分匹配,以确保组间可比性。主要有效性终点是EXCOR组和ECMO历史对照组在30天时的死亡风险比。主要安全性终点是将严重不良事件的发生率与通过文献回顾确定的器械性能目标进行比较。研究结果表明,与ECMO历史对照组患者相比,EXCOR组的死亡风险在统计学上显著降低。
通过HDE批准后,申办方对EXCOR儿科器械进行了HDE批准后研究(post-approval study,PAS),并在2017年,将原始HDE转为PMA[42]。PMA与HDE同为三类医疗器械的上市申请途径,在审批流程中的主要差异体现于对器械有效性的充分证明[47]。新的PMA申请在引用原始IDE试验数据与ELSO中RWD的基础上,利用了HDE批准后的患者使用数据辅助有效性的确定。为确保评估所有EXCOR器械使用的数据,FDA要求申办方额外收集了非HDE批准后研究患者使用EXCOR治疗的全部RWD,与HDE批准后研究的受试者、IDE试验队列和IDE同情性使用的患者数据合并,作为补充的临床证据,以全面地评估EXCOR在儿科人群中使用的有效性和安全性。结果显示,与植入EXCOR作为首个机械性循环支持治疗的患者相比,EXCOR植入前接受过ECMO治疗的患者死亡风险明显增加。基于对美国所有接受EXCOR治疗的RWD总体回顾,以及IDE试验观察到的结果,FDA认为用于原始HDE批准的研究结果在真实世界环境中具有一致性和可预测性,且具有临床意义的患者获益得到了一致的证明[46]。
综上所述,FDA可以合理地确定EXCOR提供心脏机械性循环支持的有效性。作为批准的一个条件,FDA还要求申办方建立上市后监督注册登记系统,以继续监测EXCOR在儿科人群中使用的长期有效性和安全性[51]。
2.3.3 RWE用于产品全生命周期监管示例
由某公司研发的IN.PACTTM AdmiralTM紫杉醇涂层经皮腔内血管成形术(percutaneous transluminal angioplasty,PTA)球囊导管是使用高质量RWD辅助上市前和上市后(产品全生命周期)监管决策的实例[52-53]。FDA最初于2014年批准该导管用于PTA治疗新发或再发病变的外周血管疾病,并于2016年使用来自美国血管外科学会(Society for Vascular Surgery,SVS)血管质量倡议(Vascular Quality Initiative,VQI)疾病登记系统的RWD作为同期对照组,扩展其治疗适应症至支架内再狭窄(in-stent restenosis,ISR)病变。SVS VQI是一个高质量的国家疾病登记数据库,旨在通过收集、分析和共享血管临床数据来提高血管医疗的质量、安全性、有效性和成本效益[54]。VQI疾病登记数据库收集了患者的人口统计数据和临床实践中治疗程序的详细信息,包括设备标识符、既往病史以及随访数据等[55]。通过使用网络第三方平台,全美国各地的参与医院所收集的治疗和随访数据反映了不受任何研究项目干扰的医疗标准。为了保证可靠性,特定医疗器械评估项目的数据元素、临床终点、相关数据表格的确定和采用都与多个利益相关者进行了合作,包括医疗器械行业、参与VQI的医院和医生以及FDA,以达到系统性和一致性[56-57]。为确保数据的准确性和完整性,VQI实施了多项措施,包括错误捕捉、不完整记录监控、定期的网络研讨会、随机审计和基于统计的多变量模型审计等[58]。异常数值和可疑数据均可溯源,并通过源数据文件进一步验证[57]。高质量的数据收集是通过持续的长期随访和基于医疗索赔数据的审计来确保的,以此最大限度地减少对临床结局的选择偏倚,并减少缺失数据以加强数据的完整性[55-56]。
该研究中,设备组的164名受试者均从申办方的IN.PACT全球性研究中选择,研究设计为前瞻性、开放性、单臂、观察性研究,并在美国以外的31个地点接受了IN.PACTTMAdmiralTM紫杉醇涂层PTA球囊导管的治疗。在根据预先制定的纳入和排除标准进行仔细筛选后,以153名来自SVS VQI疾病登记数据库的接受标准PTA治疗的受试者作为同期对照组。主要有效性终点是12个月时靶病变血管重建的发生率。通过严谨设计和实施的临床与统计评估,研究对20个预先指定的变量进行倾向评分调整,以平衡2个研究队列之间潜在的基线差异,并确保患者群体与结果的可比性。为充分评估有效性和安全性终点,研究设置了足够的随访时间,并进行了敏感性分析评估缺失数据的影响。此外,作为批准上市的一个条件,FDA要求申办方继续使用SVS VQI疾病登记数据库36个月,用于上市后的安全监督,监测不良事件和技术故障。FDA批准IN.PACTTMAdmiralTM紫杉醇涂层PTA球囊导管的适应症扩大,主要基于RWD相关性和可靠性的充分证明,以及科学可靠的研究设计和优效性检验,以说明该设备与RWD对照组相比的有效性[52]。
03结 论
伴随RWD/RWE领域的迅速发展,国际上对于辅助监管决策的RWE的支持取得了明显进展。全球监管机构的相应倡导和指南颁布有效地鼓励了将RWD/RWE融入医疗保健和药械监管的创新实践。最重要的是,使用适用于监管目的的RWD,是系统和广泛地使用RWE评价医疗器械产品全生命周期中有效性和安全性的关键。为了将这些概念转化为实践,全球监管机构已经认识到加快发布相关指南和框架的重要性。通过各方利益相关者的集体行动,指导医疗行业利用RWD辅助医疗器械监管决策。由高质量的RWD、严谨研究设计、稳健合理的分析方法所产生的RWE接受度将日益提高,并将在补充传统RCT的临床证据和推动监管决策的典范转移方面显示出巨大的潜力。

来源:中国食品药品监管杂志


