您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-07-14 07:05
标准物质一直是药品质量控制特别是纯度和含量分析的首选,但标准物质的供需矛盾,如有些杂质标准物质不易获取以及多个标准物质同时使用所带来的高昂检测成本等限制了标准物质的实际应用。为解决这一问题,替代方法应运而生,高效液相色谱(HPLC)校正因子法便是其中之一。
该方法最初主要用于HPLC有关物质检查时对于特定杂质的定量,进而又逐渐用于中药多指标含量的测定,发展至今已经成为一项成熟的分析方法。
近年来,随着国内仿制药一致性评价的深入开展,有关物质作为药品的关键质量属性已成为被评价的重要内容之一,而在有关物质申报资料中缺少校正因子的研究是药品审评中心通知申请企业补充完善资料的一个主要原因,这使得校正因子的研究重回企业和科研单位的视线,成为被关注的焦点。
目前,国内外有关校正因子在测定和应用方面鲜有详细的指导原则发表,因此本文综述了近年来国内外相关领域的研究进展,以期总结出校正因子在测定和应用方面的一些规律和存在的问题,给企业提供更多的参考依据。
定义
在HPLC法定量测定中,通常所讲的校正因子是某物质i与所选定的参照物质s的绝对校正因子之比,即相对校正因子,其计算公式如公式(1)。校正因子在各国药典中均有应用,只是表述方式不尽相同。
《中国药典》与《欧洲药典》(EP10.0)基本一致,使用校正因子(correction factor)的概念,差别在于国内要求当已知杂质对主成分的相对校正因子在0.9~1.1内时,可以用主成分自身对照法计算杂质的含量,超出这个范围时宜用杂质对照品或者加校正因子的主成分自身对照法计算杂质的含量;而EP10.0中则规定校正因子在0.8~1.2时仍可使用主成分对照法计算杂质含量。
《美国药典》(USP)中给出了相对响应因子(relativeresponse factor)的概念,从其各论中具体计算方法可以推导出相对响应因子与相对校正因子是互为倒数的关系,在使用时需要注意将待测峰面积(A)与校正因子相乘或与相对响应因子(f)相除以校正峰面积。

c为待测物和参比物溶液浓度
另外校正因子的使用也有一定的范围,如校正因子在0.2~5.0的范围以外时,表明杂质与主成分的UV吸收相差过大,校正因子的作用会受到显著影响,此时应改变检测波长等检测条件,使校正因子位于上述范围内,或使用结构或UV吸收与该杂质接近的另一标准物质为参照物质(如对照品易于获得、标准已采用对照品外标法定量的另一特定杂质),重新确立校正因子;如校正因子仍无法调节至适当范围,需考虑采用杂质对照品外标法等适当方法定量。
测定方法
HPLC-UV/PDA方法
单点法
制备适当浓度的待测物对照品溶液和参比物对照品溶液,分别注入液相色谱仪进行测定,按照公式1计算得到校正因子。
该方法的优点是操作简单,但由于无法排除样品处理或者检测过程所引入的偶然误差,且没有考虑到样品浓度对测定结果的影响等因素,因此该方法主要用于预实验时粗略的估计实验结果,在校正因子定值时实际应用较少。
多点法
制备适当的高、中、低三水平浓度的待测物对照品溶液和参比物对照品溶液,如果是加校正因子的主成分自身对照法计算杂质含量用,则建议待测物和参比物浓度涵盖定量限和标准限度,分别注入液相色谱仪进样测定,按照公式1计算得到多个校正因子值,求取平均值作为校正因子。目前该测定方法在中药多指标对照品替代测定中应用较为广泛,但在杂质含量确定方面应用较少。
标准曲线法
目前,校正因子的测定最常用的方法还是标准曲线测定法。具体操作如下:
精密称取待测物对照品和参比物对照品各适量,分别配制成不同浓度的系列溶液,如果是加校正因子的主成分自身对照法计算杂质含量用,则建议待测物和参比物浓度涵盖定量限和标准限度,分别注入液相色谱仪进样测定,以浓度c为横坐标,峰面积A为纵坐标,分别绘制参比物和待测物标准曲线A=kc+b,当b=0时,可以推导出两者的相对校正因子为二者斜率k之比,即公式(2):

其中k分别为参比物和待测物标准曲线的斜率。
联立方程法
前述3种HPLC方法使用的前提就是待测物和参比物最好备有色谱纯的化学试剂或标准物质,即使没有色谱纯,也要确知物质的百分含量。但在药物研发的早期,很难得到已知杂质的纯品,因此,Yu提出了一种无纯样色谱校正因子的测定方法。
根据校正因子与峰面积成正比(校正因子是常数的条件)的前提,设一“标”样仅含A、B二组分且都出峰,各组分的量之和为一定值(如A、B两组分含量之和为100%),因而可建立各组分的量之和与校正因子、峰面积三者之关联式,另一“标”样亦仅含A、B二组分,但峰面积有差异(即二组分含量有异),同样可建立三者关联式,解两个关联式即可得各组分校正因子。由此推论有几个组分就要建立相应个数的关联式求解而得到各组分的校正因子。
该方法为早期检测研发中带有多个未知校正因子组分的样品提供了一个新的解决思路,但是受早期研发样品中可能还含有很多未知杂质等因素的限制,目前可以检索到该方法的实际应用较少。
紫外吸光系数比值法
结合文献,Xu等首次推导出了公式(3),证明了两物质的相对校正因子是两者的百分吸光系数(E)之比。

因此可按吸光系数测定的相关技术要求,如对照品级别的标准物质、高中低三水平浓度测定、吸光度介于0.3~0.8之间、至少5台不同型号的UV分光光度计、2份供试液同时平行制备测定、同台仪器2份供试液的平行测定结果不超过±0.5%等,以流动相为溶剂,测定待测物和参比物在检测波长处的紫外吸收系数E1%1cm,计算比值,求得校正因子。
HPLC联用其他方法
HPLC-UV/PDA串联其他检测器方法
为解决HPLC-UV/PDA检测器方法有时很难获得待测物和参比物标准物质的难题,有研究者提出通用型质量检测器技术与PDA或UV检测器串联以确定校正因子的方法。这种通用型质量检测器产生的峰面积与相应的进入检测器的分析物的量成正比关系,而与分析物的结构特性无关,从而可以得出进入检测器中的待测物与参比物的相对数量关系,再结合UV/PDA检测器得到的峰面积,由公式(1)可推导出待测物相对于参比物的校正因子,如公式(4)所示:

在药物早期研发过程中使用此方法可以快速方便的测定特定杂质的相对响应因子,不需要进行杂质的分离纯化,也不需要杂质标准品,甚至可以不需要知道分析物的浓度。
目前可与紫外检测器串联测定相对校正因子的通用型质量检测器主要有蒸发光散射检测器(ELSD)、电喷雾检测器(CAD)、化学发光氮检测器(CLND)、示差折光检测器(RI)等。其中ELSD、CAD,其检测的峰面积与进入检测器的分析物的质量成正比关系;CLND检测的峰面积与分析物中含氮的摩尔数成正比关系。
通过这些信息可以得出进入检测器中的待测物与参比物的相对数量关系,再结合紫外吸收检测器得到的待测物与参比物峰面积,可计算出特定杂质相对于参比物的校正因子。目前仅检索到国外的相关研究工作,未见国内相关报道。
HPLC-UV/PDA检测器结合核磁定量方法
虽然紫外检测器与通用型质量检测器串联的HPLC法在测定杂质相对校正因子时方便、快速,但这些通用型的质量检测器都各有局限性。
如ELSD受精密度、线性动态范围、需要挥发性流动相等因素的局限,且ELSD的响应取决于探测器中发生的去溶剂化过程所产生的粒子的数量和性质;CLND只能测定含氮化合物,但不能够测定结构中包含—N=N—和—N—N—结构的化合物,也不能够用含氮的流动相、流动相中不能有非挥发性的缓冲盐;CAD的流动相中也不能有非挥发性的缓冲盐,而且只适用于在电喷雾电离条件下能够携带电荷的化合物,RI受灵敏度、重现性等问题限制且不能用于梯度洗脱。
NMR除具备检测器串联HPLC法的优点外,不受上述检测器的流动相、缓冲液等色谱条件的限制,q-1H-NMR检测的峰面积与分析物中相应的氢的摩尔数成正比关系,可以作为含有质子的化合物测定相对校正因子的通用质量型检测器,但前提是该化合物可溶解在合适的氘代溶剂中。于是Webster等建立了HPLC-UV与q-1H-NMR结合的方法测定特定杂质的相对响应因子(RRF)的方法。
1H-NMR检测的峰面积与分析物中相应的氢的摩尔数成正比关系,所以相对响应因子的计算公式如下:
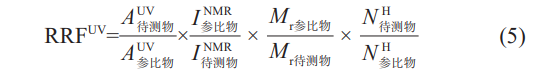
其中AUV为紫外检测器的相应峰面积;INMR为NMR谱图中相应氢的积分值;Mr为相对分子质量;NH为化合物结构中用于积分计算的相应的氢的个数。
应用
在有关物质含量测定中的应用
高效液相色谱法是目前药品有关物质检查最常规的手段,使用高效液相色谱进行杂质含量测定仍首推外标法,但该方法常常受到杂质标准物质获得和供应困难等因素的制约。
加校正因子的主成分自身对照法由于在日常检验中省去了杂质标准物质,又同时考虑了杂质与主成分的响应因子可能不同所引起的测定误差,准确度较好,目前已被广泛应用。
《中国药典》自2000年版在《高效液相色谱法》通则中增加了“加校正因子的主成分自身对照法”,同时各国药典各论也在逐渐采用该方法替代简单的主成分自身对照法,如《欧洲药典》(EP10.0)和《中国药典》(2015年版)均采用了加校正因子的主成分自身对照法检测环丙沙星杂质B、C、D、E的含量。
目前对于校正因子在有关物质检查中的应用研究大致可以分为两类。
第一类是测定特定药品杂质相对于主成分的校正因子,以完善主成分的质量标准。如Zhao等采用自建的标准曲线法测定了注射用头孢孟多酯钠中杂质A、C、D和E相对于头孢孟多酯的校正因子;Qi等建立了加校正因子的主成分自身对照法测定吉非替尼原料药中有关物质的含量,其中采用标准曲线法测定了杂质SM1、I、XV、SM2、XII、D和C相对于吉非替尼的校正因子;Guo等建立了HPLC加校正因子的主成分自身对照法同时测定琥珀酸索利那新原料药中7种有关物质的方法,其中采用标准曲线法测定了7种有关物质相对于琥珀酸索利的校正因子。
从查阅的这些校正因子测定的文献来看,目前校正因子的测定方法主要集中在标准曲线法。仅个别研究采用了多点法进行校正因子的测定,如Yan等配制了低、中、高3个浓度水平的对照品溶液,每个浓度平行制备3份样品分别进样分析,以均值作为有关物质A、B、C、D相对埃索美拉唑镁的校正因子,建立了加校正因子的主成分自身对照法测定埃索美拉唑镁原料药中有关物质含量的HPLC法。
另一类则是针对目前校正因子在测定和使用方面的不足,对方法进行改进或者开发新的方法。如针对存在多个杂质的药品使用校正因子方法时可能存在杂质定位困难的问题,Chen提出应用定向降解主成分得到相应杂质进行定位,再采用加校正因子的主成分自身对照法对该杂质进行定量的方法。
针对在药物研发的早期,很难得到已知杂质的纯品用于校正因子测定的问题,Yu提出采用联立方程组的方法测定无纯样色谱校正因子,也有人尝试两种检测器串联的HPLC法以解决这一难题,如Hong等建立了高效液相色谱法紫外检测器与蒸发光散射检测器串联(HPLC-UV-ELSD)的方法测定格列美脲中有关物质B和C的相对响应因子;Sun等采用紫外检测器和电喷雾检测器串联的高效液相色谱法(HPLC-UV-CAD)测定紫杉醇中8种有关物质的相对响应因子;Nuss‐baum等建立了紫外检测器和化学发光氮检测器串联的高效液相色谱法(HPLC-UV-CLND)测定特定杂质的相对响应因子。
针对上述质量型检测器与紫外检测器串联测定校正因子的一些局限性,HPLC-UV与q-1H-NMR结合的方法测定特定杂质的相对响应因子的方法应运而生。
Webster等在文献的基础上应用HPLC-UV-CLND方法和新建立的HPLC-UVNMR方法分别测定了扑热息痛、2-羟基喹啉、丹磺酰苯丙氨酸相对于2-喹啉酮的相对响应因子,结果显示两个方法测定的相对响应因子无明显差异;除此之外,还利用了标准曲线法、HPLC-UV-CLND法和HPLCUV-NMR法测定了酸性除草剂中3种杂质相对于2,4-D的相对响应因子,结果显示3种方法测定的相对响应因子值无明显差异。
Iqbal等利用传统的标准曲线法和HPLC-UV-NMR的方法分别对萘普生中丙烯酸杂质进行了相对响应因子值测定,证明两方法结果无显著差异。
Liu等也利用传统的标准曲线法和HPLC-UV-NMR法分别对头孢唑林中10种杂质的相对响应因子进行了测定,并对两种方法的不确定度进行了计算和比较,证明两种方法的相对响应因子值测定结果和不确定度均无明显差异。
在中药及其复方制剂多指标成分测定中的应用
目前相对校正因子方法不仅应用于药品的有关物质含量测定,而且广泛应用于活性成分特别是中药及其复方制剂中多指标成分的测定。
其实,最早将校正因子方法引入活性成分含量测定的并非在中药领域,2004年,Liu等选用稳定物质头孢唑林作为参比物质,通过HPLC方法测定该物质与力达霉素之间的校正因子,然后通过参比物质和校正因子来表征力达霉素标准品的量值,以解决不稳定标准物质量值传递的问题。但该方法并未在化学药品活性成分含量测定中推广,仅检索到个别文献应用。
中药属于多成分协同作用,随着分析技术的不断进步,其质量控制也逐渐向多指标精准测控转变。然而很多天然产物标准物质制备困难、价格昂贵,给标准物质的供应和使用单位都带来巨大的压力。
为解决这一矛盾,Wang等提出了“一测多评”的中药控制理念,即在多指标质量评价时,以药材中某一有标准物质供应的典型组分为内标,建立该组分与其他组分之间的相对校正因子,通过校正因子计算其他组分的含量。
Xie等将该方法拓展到以一种非药材中成分,但是日常检验中易获得标准物质且稳定的化合物作为替代对照品,测定替代对照品相对待测组分的校正因子,利用校正因子和替代对照品进行中药中特定成分的含量的测定,该方法在很多文献中被称为“替代对照品法”。
无论是“一测多评”法还是“替代对照品”法,两者的实质都是在没有待测成分标准物质的前提下利用校正因子结合替代对照品测定待测成分的含量。其中一部分文献校正因子的测定采用多点法,如He等以五味子醇甲为内参物,采用多点法计算了五味子酯甲、五味子甲素和五味子乙素与五味子醇甲的相对校正因子,并进行了含量计算;Lu等以人参皂苷Rg3为内标,计算了黑参中其余11种皂苷类成分的校正因子;Hou等以大黄素为内标,计算了何首乌中其余3种成分的校正因子。
另外,为了减少多点法中偶然误差对于均值带来的巨大影响,近年来也逐渐有人开始采用标准曲线法,该方法中个别点的偏差对于校正曲线斜率影响较小,且无需逐个浓度计算相对校正因子,更加简便高效。
如Liu等采用多点法和斜率校正法分别计算了元胡止痛系列制剂中4种成分的相对校正因子,两种方法的计算结果基本一致;Zhang等用斜率校正法,以绿原酸为内标,测定了甜叶菊中其他5种绿原酸类成分的相对校正因子。
目前该方法也已经逐渐从研究走向成熟应用,各国药典中也有相应收载,比如《欧洲药典》(EP10.0)收载的大蒜粉质量标准中,将大蒜辣素列为其控制指标,并采用羟苯丁酯作为大蒜辣素的替代对照品,从而解决了大蒜素不稳定的难题;《中国药典》(2015年版)一部黄连药材含量测定项采用了以盐酸小檗碱测定小檗碱、表小檗碱、黄连碱和巴马汀4个生物碱成分的一测多评方法。
问题和小结
标准曲线法校正因子的测定
相对校正因子用于杂质的定量或者中药多组分活性成分的定量方法已经得到广泛的应用,但是其在使用过程中还存在一些问题需要统一或者规范,其在中药方面的应用已经有多人对相应的技术要求进行过建议和总结,但在杂质定量研究方面相关报道甚少,因此本文将文献中相对校正因子在杂质定量方面应用存在的一些问题进行了总结。
曲线是否需要强制过原点
对于色谱方法,校正曲线的斜率b=0是一种理想的状态,事实上由于每一台仪器都有其检测限度,需要待分析物达到一定浓度水平以后才能检测出信号,以及溶剂干扰等系统误差的存在,因此截距一般不会为0。
正的Y轴截距表示高浓度响应的饱和度或响应处有干扰存在,负的截距说明可能方法灵敏度有问题,或被分析物质残留在容器或者HPLC系统中。但是标准曲线法使用斜率求取校正因子的前提是假设截距b=0,由此可见无论是否对截距进行校正,这种方法对于相对校正因子的测定都会产生一定的误差。
目前对于标准曲线法求校正因子的截距和斜率未检索到明确的要求。因此文献对于校正曲线的处理也有两种方法,一种是将截距校正为0;大部分则未做校正。但校正因子在中药方面的应用中有一些相关要求的报道,如校正曲线相关系数r不得低于0.999,k/b值大于100时,b值可以忽略不计。
Zhao等还以实例证明在中药多组分含量测定中采用截距校正后的曲线斜率求相对校正因子方法最为合理。但截距校正与否对校正因子计算会产生多大的影响,哪一种方式计算得到的杂质含量更为准确未见文献报道。
校正曲线浓度范围的选择
在进行杂质定量方法验证时,线性验证是方法验证中必不可少的一项内容,由于在验证过程中要分别建立杂质和主成分浓度和响应值之间的校正曲线,可以就此以两校正曲线的斜率计算出相对校正因子,因此很多研究对于校正曲线浓度的范围便遵从线性验证的要求从定量限到限度值的120%~150%。也有专家建议标准曲线法测定校正因子的浓度范围要涵盖定量限和标准限度。
从目前检索到的文献看,有些文献的确涵盖了定量限和标准限度,有些则仅涵盖限度或未明确定量限,其中一些文献上限定了限度水平的200%甚至500%,高浓度范围对于溶液的配制可能更为简单,低浓度范围则更加接近正常杂质检测水平,不同浓度范围所得到校正因子是否有所差别未检索到文献报道。
校正曲线上浓度点的选择
在计算相对校正因子时,多采用线性验证时的校正曲线,因此一般要求至少制备5份不同浓度的对照品溶液用于校正曲线的绘制。在配置不同浓度的对照品溶液时应尽量采用逐级稀释,在校正曲线上的点也应均匀分布,也有一些文献浓度点分布并不均匀,如在低浓度处选点更加密集,曲线上浓度分布对校正因子的测定是否产生影响未见文献报道。
涉及校正因子的方法开发与验证
目前对于杂质与主成分相对校正因子的测定多在进行有关物质方法线性验证时直接以校正曲线的斜率顺带求得,仅在《中国药典》2015年版四部《9101药品质量标准分析方法验证指导原则》中明确规定了校正因子需进行准确度验证,但对于测定校正因子的方法开发以及耐用性验证没有明确的要求。
在检索到的有关化学药品具体品种校正因子的26篇相关文献中,有16篇文献在计算相对校正因子时考虑到了仪器、色谱柱的相关影响,以在不同仪器不同色谱柱上测定得到的均值作为校正因子;有11篇文献进行了校正因子的耐用性验证,但验证项目不一,主要涉及人员、实验室、仪器、色谱柱、流动相pH值和比例、流速、试剂等级、柱温、粒径等多个影响因素,其中检测波长、色谱柱、柱温、流速都被报道过对校正因子测定结果有显著影响。
由此可见,应该在方法开发时对于特定杂质相对校正因子的耐用性根据具体情况进行全面考察,如果校正因子发生显著变化,应对具体测试条件在方法标准中加以严格限定,这样相对校正因子才能作为一个常数,直接载入质量标准用于杂质的准确定量。
校正因子的使用
杂质色谱峰的准确定位是保证校正因子法应用的前提,因此校正因子测定时多采用相对保留时间进行杂质定位,对于成分复杂样品,相对保留时间不能准确定位时,可考虑选用定性用的标准物质。
从目前查阅文献看,其中一些研究在确定相对校正因子时并未明确特定杂质的相对保留时间,提示可能对相对保留时间对于校正因子应用的重要性认识不足,但也有一些研究在考察了相对校正因子耐用性的同时考察了相对保留时间的耐用性。
由于很多色谱条件如色谱柱、柱温、流速等均会对保留时间产生影响,因此建议在使用相对校正因子结合相对保留时间进行杂质鉴别和定量时,对相对保留时间也进行耐用性考察,如果要求条件苛刻,同样需要在质量标准中进行明确规定以保证检测方法具有足够的重现性。
展望
随着大众对于药品安全性认识和需求的不断提高,药品有关物质检查中进行特定杂质控制会逐渐成为药品质控的常态,标准物质无法及时供应的情况下,采用相对校正因子计算特定杂质的方法也会越来越普遍。相对校正因子将成为药品HPLC方法中用于杂质含量测定和限度检查过程中一个至关重要的参数。
国内外采用相对校正因子方法用于杂质控制的研究已较为广泛和深入,其中许多方法被各国药典收载,具有良好的发展和应用前景。未来针对目前研究过程中存在的一些问题还需加强标准化和规范化的研究。

来源:铭研医药


