您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-07-14 22:15
杂质分析是一个老生常谈的问题了,杂质控制研究一直都是一项浩大的工程。尽管国家也出台了多个技术文件,指导研发人员进行杂质研究,但限于药品研究的复杂性,研究人员的技术水准、研发理念、对指导文件的理解等,导致药品申报中关于杂质研究的发补问题层出不穷。本文仅就化药API研究中关于潜在杂质的分析提出自己的一些看法,希望能给各位同行提供一些思路,起到抛砖引玉的作用。
杂质有多种分类方法,按照其理化性质一般分为有机杂质、无机杂质、残留溶剂,按来源又可以分为工艺杂质、降解杂质、从反应物及试剂中混入的杂质等,按毒性又可以分为一般杂质和毒性杂质;潜在杂质这种说法没有看到正式的分类规则,一般的理解是指经分析可能存在的一类杂质,属于杂质风险评估理念下的一个概念。下文根据其来源分别进行讨论:
1、 元素杂质
受地域环境影响(重金属污染),国外药品申报时对元素杂质的关注度一直都很高。近年来CDE也越来越关注元素杂质问题,如果NDA研究过程中未对潜在元素杂质进行研究,CDE大概率会就此问题发补。因此在NDA申报中,建议参考ICH Q3D,按照不同项目类型确定元素杂质的研究种类及限度。
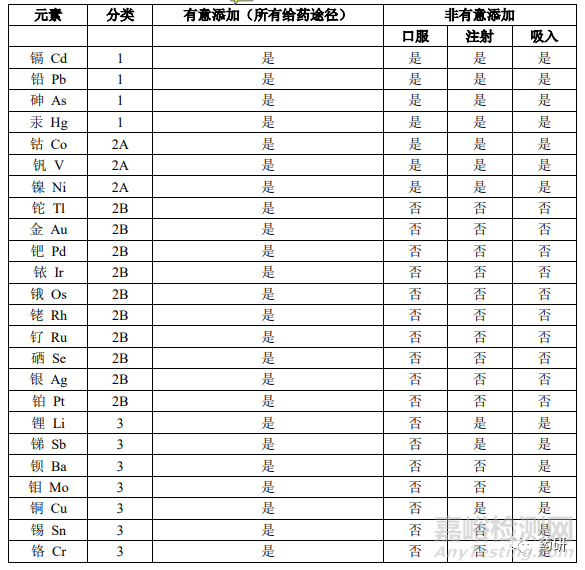
图1.不同剂型需要关注的元素杂质
元素杂质一般在终点进行控制,常用检测方法为电感耦合等离子体质谱法(ICP-MS)、原子吸收分光光度计法;ICP-MS法可以同时测定多种元素杂质,准确度高,灵敏度好,建议采用此方法。
研究过程中可将元素杂质的控制阈值设定为其PDE值的30%,如果采用稳定的生产工艺生产的多批次样品检测结果中元素杂质的水平始终低于阈值,可以认为已对元素杂质进行的足够的控制,该类潜在杂质在终产品中存在的可能性很小,质量标准中无需对该项进行控制。但是,需要多少批次样品检测结果来证明控制力足够呢?目前CDE老师也没有明确的标准。
2、 潜在工艺杂质
IND审评意见中,常见的一条就是加强本品的杂质谱研究,这里的研究必定包含对潜在工艺杂质的研究与评价。
潜在工艺杂质是在工艺过程中可能存在的杂质,包括起始原料引入的杂质及其衍生物,副反应产物,反应试剂,中间体等。此类杂质可以在过程中控制,也可以在终点进行控制。限度常用杂质清除转化研究来确定:在反应物中加入一定量的杂质,经过一步或多步反应后,該杂质及其衍生物低于检测限或关注阈值,即可证明工艺对该杂质有足够的清除转化能力;加入的量可以设定为该杂质的限度,本反应步骤可以作为该杂质的控制点。这类杂质的研究需要合成部门与分析部门紧密合作:合成部门需要结合工艺反应特点,尽可能全面的推断出需要研究的杂质(如起始物料中可能带入的杂质,反应过程中可能进行的副反应及产生的杂质),杂质结构准确,反应过程合理正确;分析人员需要确保分析方法的检测能力,使杂质在较低水平时仍能被准确检出。常用的分析方法有许多种,气相、液相、液质联用、定量核磁等,根据需要选择合适的方法。需要注意的是,近年来CDE对粗品母液的关注度较高,建议对粗品母液中的主要杂质进行研究,了解其结构、来源及消除等信息。
3、潜在降解杂质
潜在降解杂质一般通过强制破坏实验和影响因素实验来研究。
对于强制破坏实验,一般要求主成分破坏5%-15%为宜;有的主成分性质很稳定,依然要求所有破坏条件中至少有一种条件可以破坏5%以上。需要重点关注的是强制破坏实验产生的“主要”的降解产物,可以按照Alsante等人所做出的研究工作来确定哪些是主要降解产物,即:该降解物产生的量大于总体降解量的10%,并且还要大于最大单个降解物含量的25%(见图2、图3和图4)
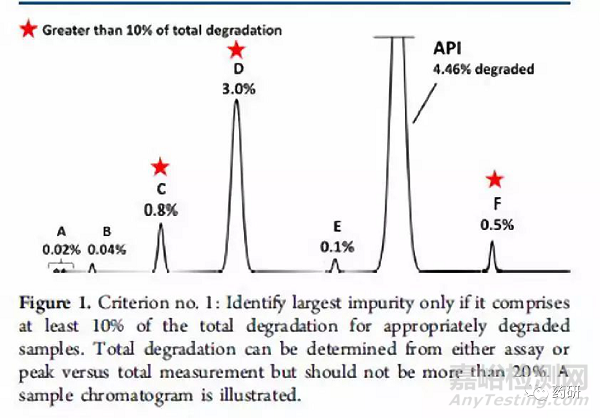
图2 主要降解产物判定示例图一
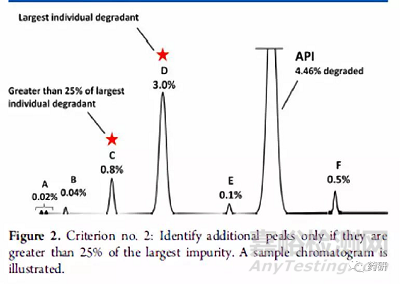
图3 主要降解产物判定示例图二
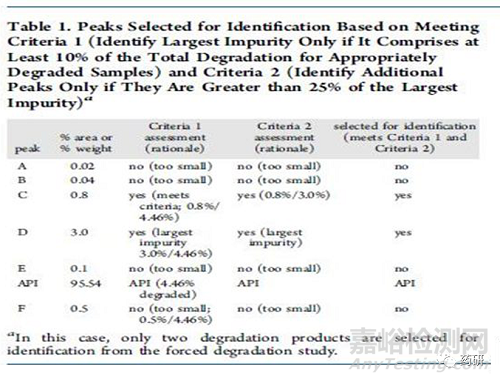
图4 主要降解产物判定示例图三
对于影响因素试验,新产生大于0.10%的杂质建议都进行研究。
另外,合成人员需要根据药品的结构特点,推断可能的降解途径及降解杂质,并将可能的降解杂质合成出来,验证该在杂质能否被成品的检测方法准确检出,以此证明成品的检测方法有足够的检测能力。目前这一研究思路很容易被忽视,NDA申报中由这一问题引起的发补较多。
需要注意的是,以上各潜在杂质的研究均未考虑杂质的基因毒性,一般在杂质结构确定后,需先进行遗传毒性评估,对于有遗传毒性的杂质,其限度设定需要先满足安全性的要求。

来源:药研


