您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-07-16 23:17
世界主要国家和地区药品附条件批准上市政策综述
Overview of Conditional Approval Polices and Guidance in Major Countries and Regions of the World
摘 要 / Abstract
为了有效应对严重且危及生命的疾病对人类健康的威胁,加快医药产品的审评审批,满足人们用药需求,世界主要国家和地区监管机构陆续出台了一系列加快药品上市注册的政策。附条件批准作为其中的一条重要路径,可使创新医药产品在尚未满足完全批准的条件时,提前获得上市批准,从而缓解亟待满足的医疗(市场)需求。本文整理了世界主要国家和地区附条件批准的历史及法律法规,并对比了附条件批准政策的关键要素,以期对我国药品附条件批准上市工作的开展提供有意义的借鉴和参考。
To address the threat of serious and life-threatening diseases or conditions to human health,expedite the development and review of new drugs,and meet public need for new drugs,regulatory agencies around the world have explored policies to speed up drug approval.As one of the important pathways,conditional approval grants marketing authorization to innovative pharmaceutical products in advance before they meet the criteria for full approval,so as to meet urgent medical (market) needs.This paper reviews the history,laws and regulations of conditional approval in major countries and regions of the world,and compares the key elements of conditional approval policies,so as to shed light on the implementation of conditional approval in China.
关 键 词 / Key words
附条件批准;加速审批;附条件的符合通知;附条件早期批准体系;临时批准
conditional approval; accelerated approval; conditional marketing authorization; notice of compliance with conditions; conditional early approval system; provisional approval
为了有效应对严重疾病对人类健康和生命的威胁,加快医药产品审评审批,使人们能够尽快用上更有效的创新医药产品,缓解急需的医疗需求,是摆在各国药品监管机构面前的一项重大课题。从20世纪末开始,世界各主要国家和地区的监管机构陆续出台了一系列的政策尝试解决这一问题。如美国FDA的快速通道(Fast Track designation)、突破性疗法(Breakthrough Therapy designation)、加速审批(Accelerated Approval)、优先审评(Priority Review designation)[1];欧洲EMA的附条件批准(Conditional Marketing Authorisations)[2]、加速审评(Accelerated Assessment)[3]、PRIME通道(Priority Medicines)[4]、特例批准(Exceptional Circumstances)[5]和中国的突破性治疗药物程序、附条件批准程序、优先审评审批程序、特别审批程序[6]等。附条件批准或加速审批作为其中的一个重要路径,可使创新医药产品在尚未满足完全批准的条件时,提前获得上市批准,从而缓解亟待满足的医疗(市场)需求。
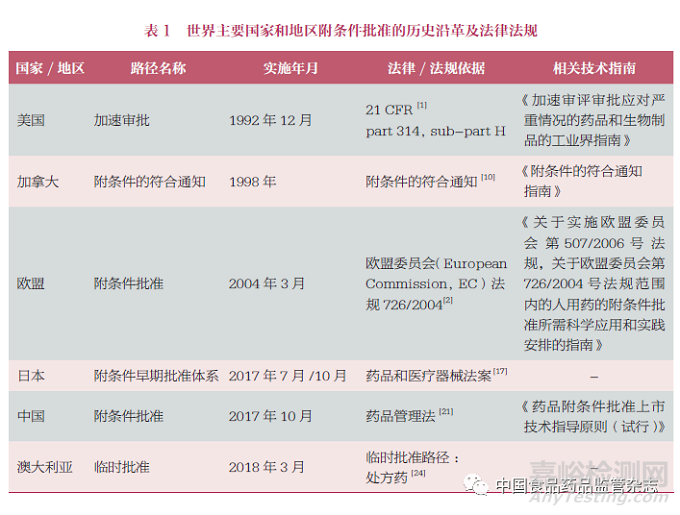
01世界主要国家和地区附条件批准的历史及法律法规
美国是世界上最早开展加速审批(对应附条件批准路径)的国家。1992年,由于人类免疫缺陷病毒(human immunodeficiency virus,HIV)传播带来的严重威胁,美国在联邦法规中加入了Subpart H,增设了加速审批路径(Accelerated Approval,AA)。在此之前,美国只有常规审批这一条路径。之后不久,生物制品也纳入到了加速审批的范畴[7,8]。2012年,《FDA安全和创新法案》(Food and Drug Administration Safety and Innovation Act)对加速审批的定义和范围做了进一步澄清和扩大[9]。2014年,FDA颁布《加速审评审批应对严重情况的药品和生物制品的工业界指南》(Guidance for Industry Expedited Programs for Serious Conditions-Drugs and Biologics),对美国加速审评审批的4条路径,包括加速审批的相关定义、范围、条件和上市后监管等做出了详细的规定[1]。
1998年,为了向罹患严重危及生命疾病的患者提供及时的治疗,加拿大卫生部(Health Canada)在《食品药品管理法》(Food and Drug Regulations)的C.08.004 中明确了附条件的符合通知[Notice of Compliance with conditions (NOC/c)]路径[10]。并于2003年和2005年对此路径进行了2次修订,以回应工业界和其他方面的请求,包括该政策的实施标准不一致,透明度需要提高,应对此路径进行更多地宣介教育等[11]。加拿大卫生部健康产品和食品司于2002年颁布了《附条件的符合通知指南》[Guidance Document,Notice of Compliance with conditions (NOC/c)],并于2005、2011和2016年多次修订[12],对附条件符合通知的相关定义、目的、范围、背景、上市所附特定条件、产品广告和说明书要求等做了详细地规定和说明。
欧盟的附条件批准始于2004年,欧盟委员会(European Commission,EC)法规726/2004,Article 14-a,7M中明确表示,在与申请人协商后,监管机构可附加某些特定义务(specific obligations,SO)后批准新药上市,该授权由监管机构每年审查一次[2]。2006年,EC法规委员会第507/2006号文件进一步定义了附条件批准(Conditional Marketing Authorization,CMA)的规定[13],并在稍后颁布了附条件批准的指南[Guideline on the scientific application and the practical arrangements necessary to implement Commission Regulation (EC) No 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004][14]。
日本的对应路径名称为“附条件早期批准体系”(Conditional Early Approval System),由厚生省分别于2017年7月(创新医疗器械)[15]和10月(药品)[16]发布通知开始实施,通知中明确规定了附条件早期批准体系的准入条件、相关定义、关键程序、沟通交流、上市所附特定条件以及产品状态变更等。该监管体系于2019年12月立法,并于2020年9月生效[17,18]。
中国的附条件批准始于2017年10月,由中共中央办公厅、国务院办公厅印发《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(以下简称《创新意见》)。其中明确指出:“(九)加快临床急需药品和医疗器械审评审批。对治疗严重危及生命且尚无有效治疗手段疾病以及公共卫生方面等急需的药品医疗器械,临床试验早期、中期指标显示疗效并可预测其临床价值的,可附带条件批准上市,企业应制定风险管控计划,按要求开展研究。”[19]2017年12月《临床急需药品附条件批准上市技术指导原则(征求意见稿)》发布,详细规定了附条件批准上市的基本条件、疗效评价、上市申请、上市后要求和撤销[20]。2019年12月1日新版《药品管理法》施行,第二十六条规定“对治疗严重危及生命且尚无有效治疗手段的疾病以及公共卫生方面急需的药品,药物临床试验已有数据显示疗效并能预测其临床价值的,可以附条件批准,并在药品注册证书中载明相关事项”[21]。2020年3月30日,国家市场监督管理总局发布《药品注册管理办法》,其中专设章节明确了“附条件批准”的范围、同时申请优先审评审批、申请程序上市后要求以及在何种情形下注销已批准文件等[6]。2020年4月,《药品附条件批准上市申请审评审批工作程序(征求意见稿)》(以下简称《工作程序》)发布,明确指出《工作程序》总体思路对应FDA的加快审批通道。对附条件批准的适用范围和认定条件、工作程序和工作要求做出了详细规定[22]。2020年11月19日,《药品附条件批准上市技术指导原则(试行)》正式发布[23],在2017年征求意见稿的基础上进行了进一步的修改和完善。
澳大利亚于2018年3月开始实施“临时批准”(Provisional Approval),颁布指南明确了临时批准的准入条件、5个步骤以及实施时间计划等,并为之配备了详细的步骤程序文件[24]。
02世界主要国家和地区附条件批准的关键要素对比
本文进一步对比了以上主要国家/地区附条件批准的关键要素,包括附条件批准的定义/目的、准入条件、上市前评价要求、上市所附特定条件等,如表2所示。



03讨 论
通过以上的总结和对比,发现世界主要国家和地区自20世纪90年代以后陆续颁布了附条件批准的相关法律法规和指南文件,其目的均是加速急需产品上市,满足人们医疗需求。对于治疗/预防/诊断有临床急需的疾病的医药产品,如果其临床试验数据显示了一定的有效性和安全性,经评估提前上市的获益大于风险时,可以批准其提前上市,并要求申请人在一定期限内完成上市所附特定条件,而后根据上市后研究的结果决定批准产品完全上市或者撤市等。
从路径命名上可以看出,美国更强调其“加速”的功能;加拿大、欧盟、日本和中国均强调了这一路径是“附条件”的批准,在获批上市之后,需要申请人继续完成上市所附特定条件;而澳大利亚更加强调了“临时”这个要素,并给予这个“临时批准”提出了6年的期限要求,如果申请人在6年之内未能证明产品上市所附特定条件要求的有效性和安全性,则这一批准将失效。
世界主要国家和地区对附条件批准的各相关要素做了详略不同的规定。从准入的适应症而言,美国只有一个条件为“严重或危及生命的疾病或情况”,加拿大为“严重,危及生命或严重衰弱的疾病或情况”;欧盟包含三种情况:“严重或危及生命的疾病”“公共卫生急需”和“被认定为罕见病药的医药产品”;日本有两种情况:“严重疾病”和“被认定为罕见病的医药产品”;中国有三种情况:“严重危及生命”“公共卫生急需”和“重大突发公共卫生事件”;澳大利亚只有一种,为“严重情况”。即针对“严重危及生命”的疾病的医药产品,在以上各国均可以申请附条件批准;针对“公共卫生急需”的医药产品可以在欧盟和中国申请附条件批准;而欧盟和日本允许罕见病药品申请此路径。在其他的准入条件里,各主要国家和地区均强调了“未被满足的医疗需求(无有效疗法,或此疗法比原有疗法有突出优势)”,日本还增加了一条“确证性试验难以开展或者因为病人数量少而需要很长的时间”。可见,日本的准入条件较其他国家和地区严格。另外,加拿大和澳大利亚都明确规定了只有新药和新增适应症可申请此路径。
对于上市前评价的要求,美国强调了“和现有疗法相比有明显优势”以及“基于替代终点和中间临床终点显示的疗效可以预测临床获益”;加拿大要求产品有“较确定的临床获益”、“在获益风险评估的基础上,安全性可接受”和“高质量”,并明确指出产品的疗效可基于替代终点、Ⅱ期临床试验或者单臂的小到中型Ⅲ期临床试验;欧盟更加强调“获益大于风险”,申请人后续可以提供“完整的数据”;日本要求比较简单:“临床试验不一定是确证性临床试验,显示了一定程度的有效性和安全性”即可;我国更加侧重于“基于替代终点、中间临床终点或早期临床试验数据”可以显示疗效并预测临床价值,并强调了附条件批准在“重大突发公共卫生事件”中的应用;澳大利亚要求上市前数据显示产品有“突出的治疗优势”,而这一结果的得出可以基于替代终点、单臂试验、非随机对照试验、中期分析、小的数据库和亚组人群。因此可以看出,对于附条件批准的上市前关键研究,各国药监机构没有明确规定必须是Ⅲ期随机对照试验,有的国家甚至明确表示可以是Ⅲ期基于替代终点的单臂试验。而对于其效果评价,有的国家强调需要获益大于风险,如加拿大、欧盟和中国,有的国家强调其疗效是基于替代终点或中间临床终点的,如美国、加拿大、中国和澳大利亚。
对于上市所附特定条件,美国、加拿大和澳大利亚均强调了“确证性试验”为第一要务;欧盟的“特定义务”可以是干预性试验也可以是观察性研究,日本明确表示上市后研究可以是上市后监测数据和注册研究。欧盟和日本上市后要求的标准较低,这和其准入条件较高也是相关的。美国指南规定上市后确证性研究通常应该使用临床终点,但是使用替代终点更长时间的试验以及不同人群的试验亦可接受;而我国的“上市后临床研究计划”要求,根据替代终点和早期临床试验数据而附条件批准上市的产品,应设计并完成以临床终点为主要终点指标的确证性临床试验;根据中间临床终点而附条件批准上市的产品,应继续完成确证性临床试验。可见我国对上市后试验的研究终点比美国有更高的要求。在上市后研究之外,各国家和地区还有一些额外的上市后要求,如,美国要求申请人在获批上市前提交说明书和广告;加拿大要求提交上市后年度进展报告、产品针对病人和医护人员的教育材料、说明书和广告,并加强上市后监测;欧盟要求定期提交安全性报告和年度更新;日本要求为医疗机构和其他机构提供产品使用指南;我国要求药品说明书明确产品为附条件批准和加强上市后风险管理计划等,大多集中在安全性和宣传要求上。
综上,本文对比了世界主要国家和地区附条件批准的政策,发现其核心要求基本一致,但是在具体的细节要求上,各国家和地区各有侧重,产生这一差别的原因可能与该国家和地区颁布政策的时代背景、创新制药产业的状态、国家政治体制、监管理念等相关,还需要进一步的研究。另外,世界各主要国家和地区附条件批准的实施是否和政策要求相一致,以及各主要国家和地区附条件批准的监管实践有何异同之处,值得进一步研究。
引用本文
张晓方,朱枫,武阳丰*.世界主要国家和地区药品附条件批准上市政策综述[J].中国食品药品监管,2021(12):79-86.

来源:中国食品药品监管杂志


