您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-07-19 07:24
近年来,细胞治疗产品作为创新生物制品中的明星,在国内呈现爆发式增长,以 CAR-T 细胞产品为例,从 2016 年到 2020 年,在中国临床试验注册中心注册的临床试验从 27 项增长到 357 项[1]; 截止2021 年底,全球已上市的 7 款产品中,我国占到了 2款。在这类热门创新品种的药学研究中,其特殊的生产工艺和质量属性,对于传统的质控方法提出了挑战和更严苛的要求。以无菌质控为例,因细胞产品无法进行终端灭菌或除菌过滤,对工艺的无菌保障和过程控制要求更高,而目前研发/上市最多的自体型 CAR-T 细胞治疗产品,又存在个体差异大、单批产量少、部分产品效期短、临床需求紧迫,急需快速放行。但是经典的无菌检查法,需要耗时至少两周,成为制约放行时间的最大短板之一,其采样方案又按照大规模生产场景设计,已无法满足这类产品的过程控制和成品放行需求。
为了适应新品种新形势,指导这类产品的药学研究以及日常的产品质量控制,国家药监局从多渠道相继发布了《细胞治疗产品研究与评价技术指导原则( 试行) 》、《CAR-T 细胞治疗产品质量控制检测研究及非临床研究考虑要点》、《免疫细胞治疗产品药学研究与评价技术指导原则( 征求意见稿) 》等一系列指南,其中鼓励开发和验证、应用快速 /微量的新方法用于微生物质控放行,并将过程控制和放行检验结合,以满足细胞制品特殊而迫切的要求[2-4]。2021 年 10 月 26 日,国家药典委官网 公 示 了《细胞类制品微生物检查法》草案[5],本文从背景,注意事项等方面,对这类方法的应用规范进行浅析。
1、国内外标准进展情况结语与展望
各国药典所收载的经典微生物方法已有数十年历史,其基本原理依赖于人工判读培养基中污染微生物的自然恢复生长,方法虽然久经考验,但是与飞速进步的理化方法相比,试验周期冗长,不论在生产过程控制还是在成品放行中都成为制约因素。为了弥补经典方法的短板,需要引入新的替代/快速方法,各国药典也与时俱进地推出了一系列指南,除了适用于各类药品的方法引进/评估/验证/应用的通用原则外,还为创新生物制品配套了专用的方法和放行策略。
1. 1 替代/快速微生物方法引进/评估/验证/应用的一般性原则
替代/快速微生物方法往往源自食品、临床、环境等其他应用领域,而药品的微生物质控项目通常属于重要的安全性指标,因此,为了保障用药安全,需确认新方法的可靠性,必须经过药典体系下严谨的评估和验证。
为了在制药领域引进更新更快速的微生物检测方法,各国药典或行业协会在 2000 ~ 2012 年期间相继新增了相关指导原则,包括美国注射剂协会( PDA) 第 33 号 技 术 报 告,美 国 药 典 ( USP) 通 则1223,欧洲药典( EP) 通则 5. 1. 6,《中华人民共和国药典》( 简称《中国药典》) 通则 9201,这些指导原则设计为可通用于所有药品场景,为选择、开发或引进、验证和应用微生物替代方法提供了流程指导,包括应用原则、方法学参数的验证要求; 而在 2013 年后,由于创新制品和新技术的不断涌现,为了鼓励并规范新方法的推广,USP、EP 和 PDA 技术报告又进行了多次修订,在技术原理、基于风险的 4Q 确认流程、验证的统计学要求方面提供了更详细的指南,《中国药典》通则 9201 也正在修订中[6-9]。
1. 2 适用于创新生物制品的替代方法和应用原则
创新生物制品在应用替代/快速微生物方法时,除一般性原则外,还需考虑其与传统品种的差异,以选择可靠的方法,并结合生产工艺和质量属性的特殊性在验证中增加要求,设计加速放行的策略。
EP 通则 2. 6. 27 《细胞制品的微生物检查》自2007 年收载以来已经多次修订,该通则主要介绍基于培养生长信号的自动化方法,但未限定具体原理;考虑细胞制品的特殊性和局限性,在取样方案,质控菌株上的规定均与通则 2. 6. 1 的经典无菌检查法有所差异,并建议了更灵活的培养温度和培养基组合。该通则也允许采用其他方法,例如不经培养直接检测的方法,或者结合预培养和直接检测的方法,但这些方法需要按通则 5. 1. 6 的原则并结合通则2. 6. 27 对于细胞制品的建议进行方法学验证和方法适用性试验[10]。
美国 FDA 于 2012 年修订了关于生物制品无菌标准的联邦法规 21CFR. 610. 12 章节,虽然未给出具体 方 法,但 从 试 验、试 验 要 求、书 面 程 序、取样、方法确认、复试程序、记录和例外方面做了原则性规定,允许采用培养或者非培养的方法,鼓励采用更先进和适用的检测技术[11]。为了适应创新生物制品的新形势,USP 于 2019 年 12 月正式发布通则 1071《用于无菌短效期产品放行的快速微生物检查法———基于风险的方法》,适用于配药无菌制剂、正电子发射断层扫描( PET) 产品以及细胞和基因治疗产品这类短效期产品,推荐了 6 类有应用前景的快速微生物检查方法,并提出了基于风险的微 生 物 监 测 和 放 行 理 念[12]。随 后 USP 于2020 年在其药典论坛上发布了通则 72《基于呼吸的短效期产品放行用快速微生物检查法》和通则73 《基于 ATP 生物发光的短效期产品放行用快速微生物检查法》两个公示稿,分别针对呼吸信号原理的方法和 ATP 生物发光原理的方法,在采样方案,质控菌株等方面的规定均与通则 71 的经典无菌检查法有所差异[13-14]。
2、《中国药典》细胞类制品微生物检查法的拟订
国家药典委于 2019 年立项了“无菌快速检查法”( 课题编号 2019S11) 国家药品标准提高课题,课题组选择呼吸信号检测技术作为评估对象,包括二氧化碳显色、二氧化碳荧光和顶空压力信号,覆盖了主流的仪器平台,参考《中国药典》通则 9201 和国外指导原则[6-10,12-14],对 3 种主流的仪器方法与经典无菌检查法完成了比对验证及评估[15]。根据研究结果,借鉴国内外相关标准经验,完成了公示稿草案的拟订。下文将就制订草案时的考虑要点逐一进行介绍。
3、应用细胞类制品微生物检查法时的考虑点
3. 1 基本原则
本法针对的细胞治疗产品未经最终灭菌工艺处理,也不同于采用无菌生产工艺并进行除菌过滤的品种,并非传统意义上的无菌制品,参考国外药典经验[10,12-14],定名为微生物检查法而非无菌检查法。
在检测方法选择上,目前收载的呼吸信号检测技术是应用较多的方法,其采用自动化的仪器实时监控微生物培养的呼吸信号,原为临床检测开发,主要用于对血液等体液样本的快速微生物检测,也应用于临床干细胞库检测,该类技术也为欧美药典所推荐[10,12-13]。
与经典的无菌检查法相比,本法在适用场景上,
主要针对采用通则 1101 无菌检查法时具有局限性的品种。在实施原则上,由于细胞治疗产品工艺特殊,相对于传统品种无菌保障风险更高,所以务必注意产品放行大前提是基于生产工艺和无菌保障水平、微生物污染风险、使用者获益/风险等因素综合考虑的结果,而不能仅依赖检查法本身。此外,在引入这类方法前,应按通则 9201 的要求通过方法学验证。
表 1 比较了本方法与经典无菌检查法的主要异同之处。
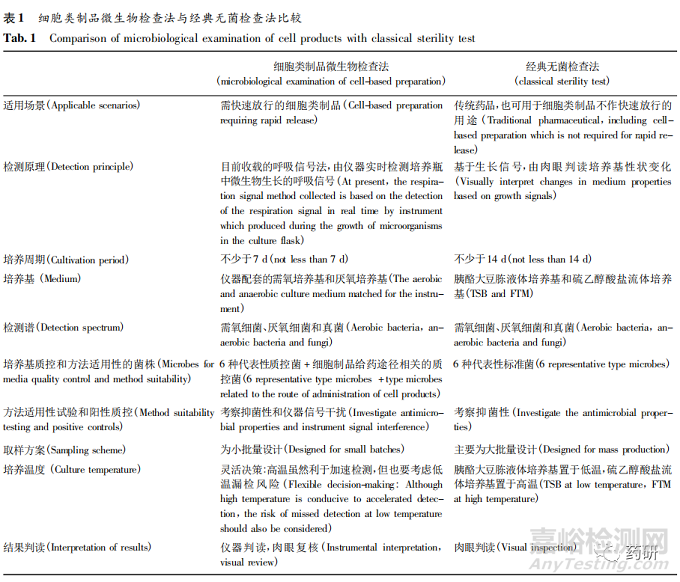
3. 2 培养基
呼吸信号的方法一般采用仪器配套的培养基,在培养基的质控方法上,也参考通则 1101 的原则操作,但是在灵敏度试验的菌种方面,需参考“方法适用性试验”章节下的规定,菌种类型较通则 1101更多。
3. 3 方法适用性试验和质控菌种
方法适用性试验是指替代方法按通则 9201 通过验证后,针对应用场景,也就是具体的细胞产品,进行产品加标试验菌的回收试验,用于考察产品成分对检测的干扰。而如果实验室在按通则 9201 开展验证期间,已采用产品加标试验菌的形式完成了各个方法学参数的验证,则可不必再重复该品种的方法适用性试验。
与经典方法的人工判读不同,本法为仪器方法,在试验中考察的干扰因素除了产品的抑菌成分外,可能还有产品成分对仪器的影响,例如对显色或荧光信号的干扰而造成读数异常。
这类方法的试验菌除了沿用经典方法的 6 种试验菌株外,还参考欧美药典经验[10,13],根据课题组试验结果[15],增加了微球菌( Micrococcus sp. ) 、酿脓链球菌( Streptococcus pyogenes) 、痤疮丙酸杆菌( Propionibacterium acnes,现 改 名 Cutibacterium ac-nes) 3 种菌株,这也是考虑到细胞产品的起始材料从人体采集可能引入的潜在污染风险的情况[16]。其中的痤疮丙酸杆菌为人皮肤上的优势菌群,为生长缓慢的厌氧细菌[17],如采用通则 1101 无菌检查法收载的硫乙醇酸盐培养基,接种量 < 100CFU置于 30 ~ 35 ℃ 培养,往往需要 6 d 以上检出,这对于呼吸信号方法也是一种检出时间上的挑战[15]。
除了上述标准试验菌外,用户实验室还可根据环境采样和历史污染情况,酌情增加相应的试验菌。
3. 4 供试品的检查法
3. 4. 1 取样方案
自体细胞治疗产品产量极少,只有数十至数百毫升,生产工艺与大批量无菌灌装的传统制品差异极大,且成本昂贵; 而经典无菌检查法的抽样方案为大批量制品设计,无法适用于细胞治疗产品的采样。因此参考欧美药典经验[10,13],本法设计了检验量要求,但需注意的是欧美药典中规定的检验量为所有培养基接 种量的总和,而目前公示稿适当从严,规定的检验量为单种培养基接种量。
在取样点上,如成品取样不适用,应考虑选择有代表性的替代取样方案,例如从所处理细胞最后接触的液体中取样[10]。
3. 4. 2 检查操作
供试品的处理和接种过程应遵循无菌操作规则,检验环境要求与通则 1101 无菌检查法一致,供试品接种量与培养基的比例应符合方法适用性试验确定的条件。
3. 4. 3 阳性质控
沿袭《中国药典》对安全性项目从严控制的传统,本法与通则 1101 无菌检查法一致,保留了阳性对照的要求,同样采用产品加标对照菌的方式操作。阳性对照菌建议选择在方法适用性试验中,发现干扰较为明显的菌种。
由于本法为仪器方法,并采用仪器配套的商品化培养瓶,因此阳性对照也是判读仪器稳定性的质控。在方法适用性试验确立阳性对照时,应评估仪器对阳性对照瓶报告阳性时间的合理范围,以建立日常工作的质控范围。
3. 5 培养条件和结果判断
3. 5. 1 培养时间
参考课题组试验结果和欧洲药典通则 2. 6. 27 的经验,公示稿目前要求培养时间不少于 7 d[10,15]。如发现生长特别慢的微生物,或在方法适用性试验中发现产品干扰难以完全消除的情况,也可酌情延长培养时间至 14 d。
3. 5. 2 培养温度
在各国药典的经典无菌检查法中,往往将培养厌氧细菌兼顾需氧细菌的硫乙醇酸盐流体培养基置于 30 ~ 35 ℃ 的高温区,将培养真菌和需氧细菌的胰酪大豆胨液体培养基置于20 ~ 25 ℃ ,以期覆盖高温细菌并兼顾部分可能低温生长的真菌。
呼吸信号方法来源于临床检测领域,为了加速培养适应人体体温的寄生微生物,一般将仪器配套的厌氧培养基和需氧培养基均置于高温生长,可较快获得阳性结果。试验结果[15]以及相关报道也表明[18],在大多数情况下排除仪器检测与肉眼观察的因素差异外,高温培养可明显加速污染微生物的生长,确实更有利于实现快速方法快速检出的设计初衷。
但是,在 GMP 生产环境中也可能出现偏好低温生长的真菌,例如青霉属,在高温培养条件下反而回收不利。虽然真菌污染在 GMP 生产环境中检出污染的几率相对较小[19],而其中偏好低温真菌的污染比例可能更低,但如出现真菌污染也是一个不易消毒清洁的风险点。因此,实验室应考虑生产工艺的无菌保障水平和污染风险,评估是否需要增加低温培养或采取其他的补充检验措施[18,20]。
考虑上述因素和国外标准经验,公示稿目前对培养温度选择做了较灵活的提示,以供不同用户实验室选择决策。
3. 5. 3 培养观察和结果判读
本法虽然为仪器方法,人为判定干扰较少,但是仪器方法本身也可能存在一定的假阳性或假阴性几率,因此为确保用药安全,在结果判读时,也需要进行目视观察: 当两种方式结果一致时可直接做出判读; 而当两种方式结果矛盾时,应按照公示稿的提示进行后续确认试验。
4、小结
细胞治疗产品为微生物污染风险较高的产品,原因在于: ( 1) 其涉及生产物料繁多; ( 2) 起始材料采集的无菌控制有限、生产流程涉及上游质粒和病毒等载体制备、临床到 GMP 生产场地往返的细胞制备; ( 3) 细胞供体本身可能带有病原微生物; ( 4) 生产中又缺乏除菌工艺。同时又由于临床需求急迫、效期短、产量小、成本昂贵,因此在质控上需要采用更快速灵活的方法。
正如传统制品的无菌性不能依赖于无菌检查,细胞制品的无菌保障也绝非依赖于快速/替代微生物检查法,而检查法的结果报告时间亦不能理解成等同于产品的放行时间。
细胞治疗产品无菌放行的决策源于无菌保障工艺的设计、验证和实施; 源于对从起始物料开始对外源污染风险的控制; 此外,除了成品的微生物检测和实时读数外,还可根据工艺特点,在过程控制中将质控点前移检测,这些工作配合使用者的获益/风险考虑,有利于做出最终放行的决策[21],同时也可以将快速/替代方法或平行实施的经典无菌检查法继续培养至14d,如发现污染,应及时鉴定并通知临床医生,为患者及时采取有效治疗措施提供依据。
本公示稿收载的方法为目前较多采用的呼吸信号原理,这类原理的方法与经典方法比较的验证案例在国外已有较多同行评议的文献报道,涵盖国外主流的仪器平台[13]; 而国内目前公开发表的文献还相对较少,国内实验室积累的经验较少,类似原理的国产仪器也在研发中,考虑到涉及安全性质控,因此在当前阶段,在应用该类仪器方法前,用户实验室应参考通则9201的要求对方法进行验证后,再参考公示稿的要求应用。
除了基于培养生长的呼吸信号方法,核酸扩增方法、ATP 生物发光方法等直接检测技术,或者将这些直接检测技术与培养方法结合,也是值得关注的候选技术,有理由期待会有更多改进和创新的微生物替代方法进入药典体系。
除了细胞治疗产品,在传统的生物制品领域,例如高风险的疫苗、高附加值的重组药物,这些品种也存在生产工序多,生产周期长,起始物料复杂,反应条件温和,易于污染微生物的情况。因此,国外已有将快速/替代微生物方法应用于疫苗等传统生物制品过程控制和成品检定的报道[22]。它能实现对微生物污染的近实时检测,掌握工艺现状并缩短等待时间,在更好地监测工艺过程的同时降低损失风险,也将成为国内推广快速无菌检查方法的潜在领域。

来源:中国药品标准杂志


