您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-08-07 17:30
摘 要 Abstract
我国自2015 年启动药品审评审批制度改革。2015 年《关于改革药品医疗器械审评审批制度的意见》和2017 年《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》明确了顶层设计,为理念转变和机制创新指明了方向。近年来,我国全面引入了全球通行的药品研发与注册技术要求,推动了我国药品审评审批制度改革创新,我国医药创新在社会经济发展中的战略性更加显著。实现研发、注册与审评环节的同步,旨在推动创新药在我国的研发与全球进度实现一致,将促进我国进一步融入全球同步研发体系,推动行业可持续、高质量发展。本文基于过去数年药品监管改革促进医药创新的改革成果,全面阐述推进创新药同步研发、注册与审评对构建医药创新生态系统的重要意义。深入分析当前同步研发、注册与审评面临的深层次挑战,聚焦“临床研究”和“监管审批”两个主题,提出10 条核心建议。全文分三期,本期围绕推动同步研发、注册与审评的背景、当前差距及关键维度进行综述,后续将从注册监管的科学性、临床研究的高效性以及监管和临床能力建设等方面分别展开相关研究。
Drug review and approval system reform has been initiated since 2015. The Opinions of the State Council on Reforming the Review and Approval System for Drugs and Medical Devices in 2015 and the Opinions on Deepening the Reform of the Review and Approval System and Encouraging Innovation of Drugs and Medical Devices in 2017 laid out the overarching design of the innovation system, and pointed out the direction for transformation of mindset and innovation in mechanism.In recent years, China has comprehensively introduced the technical requirements for drug R&D and registration that prevail globally, promoting China's reform and innovation in drug review and approval system and highlighting the strategic role of China's pharmaceutical innovation in social and economic development. The purpose of simultaneous R&D, registration and review is to help the R&D of innovative drugs in China to keep pace with global development, drive China's further integration into the global simultaneous R&D system, and enhance the sustainable and high-quality development of the industry. Based on the achievements in pharmaceutical innovation driven by the drug regulatory reform over the past few years, this paper comprehensively elaborates the significance of promoting simultaneous R&D, registration and review of innovative drugs to build a pharmaceutical innovation ecosystem. It conducts in-depth analysis of the current deep-seated challenges posed to simultaneous R&D, registration and review, and proposes ten core recommendations on the two focus areas of clinical research and regulatory approval. The research consists of three papers. This paper gives an overview of the background, current gaps and key dimensions of promoting simultaneous R&D, registration and review. Subsequent papers will focus on science-based registration supervision, efficient clinical research, as well as the regulatory and clinical capacity building.
关键词 Key words
医药创新生态系统;同步研发;监管体系建设
pharmaceutical innovation ecosystem; simultaneous R&D; establishment of the regulatory system
一、推动同步研发、注册与审评的重要意义
2015~2020 年,加入国际人用药品注册技术协调会(ICH)和多项药品审评审批制度改革政策的出台,为推动新药同步研发、注册与审评上市奠定了基础。在此历史背景下,进一步缩短我国新药上市同全球的时间差,实现患者获益、推动创新体系升级、促进产业可持续发展三方面的重要意义,需要更加科学的监管要求和更加高效的临床研究流程。同时,新药研发、注册和审评全过程需要药监局、科技部、卫生健康委、知识产权局、医保局、海关等多个政府部门参与管理,政府部门之间、政府部门与企业和研究者之间的通力协作配合,对于实现同步研发、注册与审评至关重要。
(一)背景:审评审批制度改革推动新药加速上市,使创新药全球同步上市成为可能
自2015 年以来,国家先后发布系列审评审批制度改革重要文件,为加速推动药品监管立法改革指明方向。临床试验审批时间曾是严重制约新药审批速度的环节,2018 年出台的临床试验60 天默许制为提升临床试验效率带来了质的飞跃。2019 年第十三届全国人大常委会审议通过新修订的《药品管理法》,从法律层面固化药品审评审批制度改革成果。2020 年新版《药品注册管理办法》中明确规定4 个新药上市注册加快通道,包括突破性治疗药物、附条件批准、优先审评审批和特别审批。
2017 年原国家食品药品监督管理总局加入ICH,2018 年国家药监局成功当选为ICH 管理委员会成员并于2021 年连任管理委员会成员。加入ICH 并成为管理委员会成员,大力推动了我国药品注册标准的科学化发展,加快了药品注册技术要求与国际要求的协调和统一,为我国同步研发创造了关键条件,是实现全球同步研发、注册与审评的重要基础。
从结果上看,过去五年药品审评效率提升成效显著。2016 年开始,我国药品注册申请受理量逐年上升,在2018 年更是实现了51.7%的增长。2019 年以来,药品审评解决了前些年遗留下来的积压问题,审评任务完成量增长了30%。到2020 年,药品注册申请审结任务整体按时限完成率达到94.5%,2020 年7 月至今一直保持在95% 以上。2020 年国家药监局药品审评中心受理了临床默示许可1618 件,按时限完成率99.9% ;2021 年1~4 月临床默示许可按时限完成率100%,平均审评用时已由2015 年的16 个月压缩至50 日。我国创新药临床研究和注册上市申请获批时间得到大幅改善(图1);我国近年获批创新药(包括1 类创新药和境外原研药)数量增加显著,已接近发达国家和地区水平(图2)。
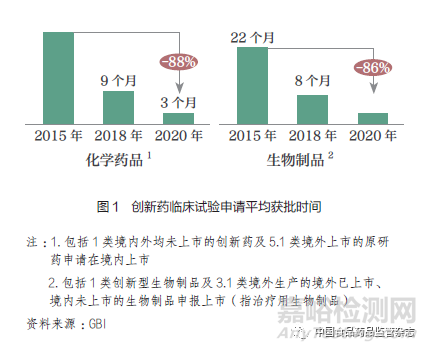

(二)当前差距:我国研发、注册与审评与全球尚存的“时间差”
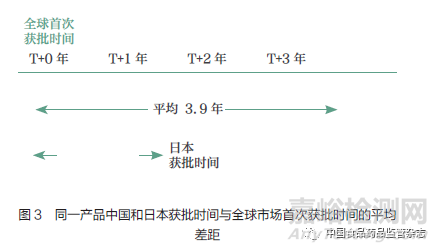
药品审评审批制度改革的“组合拳”已将新药审批时间大大缩短,使得创新药实现境内外同步注册上市成为可能。在此背景下,依然存在差距。对于2020 年我国首次获批上市的近30 个境外生产原研药(不包括再注册、原研药获批前国内已有仿制药上市以及同一产品新增适应症的情况),同一产品我国获批时间与全球其他市场首次获批时间相比,平均晚了3.9 年(30 个产品的中位数);针对2020 年在日本获批上市的跨国药企原研药分析显示,同一产品日本获批时间与全球其他市场首次获批时间的差距平均只有1.2 年(图3)。我国获批时间的差距,一方面是由于我国药品审评审批制度改革提速的成果在2020 年获批药物上还未完全显现,另一方面是由于我国仍在充分加入全球同步研发的过程中,导致在我国递交新药申请(newdrug application,NDA)的时间较晚。分析显示,在2018 年开展的全球多中心临床试验中,有19.7% 的试验纳入了日本,而同时期仅有9.4%的试验纳入了中国(图4)。

以全球同步为导向的中国监管体系的国际接轨和临床能力的全面提升,对于引领本土创新的全球化至关重要。而对于跨国药企,中国参加全球同步研发是在我国实现同步递交申请与获批的关键。回顾过往几年的行业实践,我国参与全球同步研发注册在取得成效的同时,也面临着诸多挑战。
中国外商投资企业协会药品研制和开发行业委员会(R&D-Based Pharmaceutical Association Committee,RDPAC)在2020 年完成的一项调查显示,自2017 年1 月至2020 年10 月期间,会员公司仅有8.5% 的NDA 递交(总共17 个项目)得以实现同步递交(这里的同步递交是指我国的NDA 递交发生在全球首个市场批准之前),其中多数为产品适应症扩展或新的复方产品,真正实现创新药首个适应症同步递交的情况还较少。跨国公司实现创新药全球同步递交,通常要提前4~5 年开始布局,对于2020 年前同步NDA递交的产品来说,意味着跨国公司需要在2015 年或之前就把中国放到全球研发和注册的考虑范围内。而2015 年我国药品审评审批制度改革开始前的药品注册时间长,注册申请大量积压,跨国公司需要有相当的前瞻性才可能把中国放到全球同步研发和注册中。未来同步递交产品的比例会不断上升。
(三)实现同步研发、注册与审评的重要意义
缩短尚存的差距,实现同步研发、注册与审评对于创新药在国内外加速上市造福全球患者,我国医药创新体系国际接轨互认升级,以及推动行业可持续发展具有重要意义。
1. 有助于创新药在国内外加速上市造福全球患者
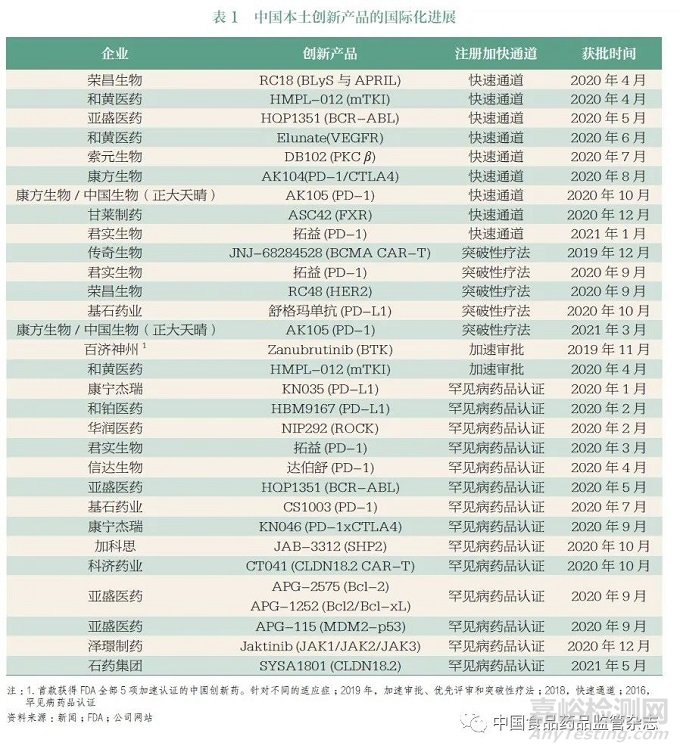
我国患者基数大(即临床资源)是助力提升全球研发效率的一大优势。实现全球同步临床研究,有助于加速创新产品在我国的研发和注册,尽早让我国患者同步受益于全球创新成果。得益于过去五年以来的一系列监管制度改革与研发体系升级,创新药在中国获批的速度和数量取得显著进展,提高了我国患者创新药物的可及性。然而,我国仍有巨大且未满足的临床需求,以恶性肿瘤为例,尽管我国患者5 年生存率已经从10 年前的30.9% 提升到目前的40.5%,但同美国近70% 相比仍有较大提升空间(图5)。同步研发、注册与审评将帮助我国患者更早获得创新药物,结合临床上的正确应用和创新药的支付保障升级,最终将提升药物可及性和患者获益。同时,我国本土创新成果尽快走出国门,可惠及更广泛的全球患者,推动构建人类卫生健康共同体(表1)。

2. 有助于我国医药创新体系能力提升,进一步融入全球创新系统
同步研发、注册与审评对监管政策的科学性、制度的完善性、审评能力和透明度,以及临床研发关键流程与环节的合理性等方面均提出了更高的要求。相关工作的推进将有助于促进我国整体医药研发体系建设,包括推动我国与国际标准接轨、优化监管审批、提升临床研究能力等。将我国纳入早期开发阶段,实现新药在我国的同步递交和同步上市,有利于发挥我国在全球研发体系中的重要作用。
3. 有助于提升整体研发能力,推动创新产业的可持续发展
考虑到创新药行业高投入、高风险的本质,同步研发、注册与审评将帮助我国本土创新药企在更短时间内与全球范围共享创新成果,并形成投资回报的良性循环,推动行业的可持续发展。对于跨国药企而言,将我国更好地整合到全球研发中,无需开展单独为我国监管注册而进行的临床研究,可以更高效地投入研发资源。
(四)大力推动同步研发、注册与审评的关键维度
当前在我国实现创新药的全球同步研发、注册与审评主要有3 条潜在路径(图6)。
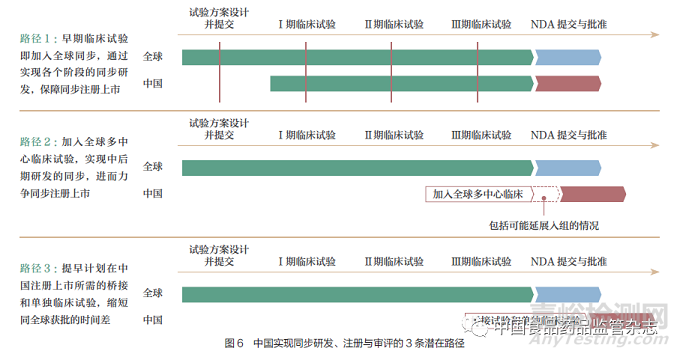
路径1 :基于ICH E17 的理念,创新药研发企业在药物全球研发早期临床试验阶段就将中国纳入,并及早与我国药品监管部门就新药研发过程中的关键问题和环节进行沟通交流,通过实现临床研究各阶段的同步,实现在我国的同步注册上市。
路径2 :基于ICH E17 的理念,创新药研发企业在药物全球研发中后期确证性临床试验阶段将中国纳入,确保获得充分的数据(包括在必要时延长中国入组时间),支持在中国的同步注册上市。
路径3 :在中国无法参与国际多中心临床试验的情况下,提早计划在中国同步注册上市所需的桥接试验和单独临床试验。需要注意的是,单独临床试验将带来药企研发费用的增加和研发周期的延长。
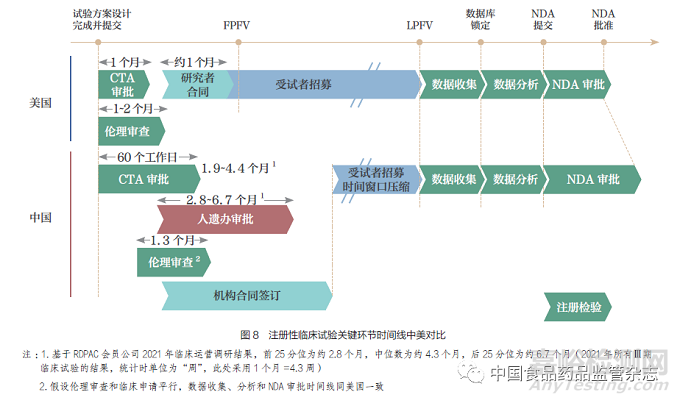
基于对3 条潜在同步路径的梳理,提升注册监管科学性和临床研究高效性是大力推动同步的关键。我国和全球的同步研发注册,要求各个监管部门、企业和研究者通力配合,在研发和注册各关键节点实现同步,单一环节的推迟都潜在影响整体研发注册进程。目前,对比全球医药研发第一梯队的美国,我国在临床试验启动阶段的时间线明显延长,整体流程优化有待提升。对于早期临床试验加入全球同步(路径1)而言,由于试验启动所需时间较长造成几乎没有招募时间窗口,我国往往因此错失加入机会(图7);而对于同步加入全球确证性临床试验(路径2)而言,我国招募时间窗口通常较短且存在较大的不确定性,导致申办方对于能否保证全球临床试验时间线产生顾虑,在一定程度上造成我国未能充分加入全球同步研发(图8)。更加科学的监管要求和更加高效的临床研究流程,有助于保证时间线上我国和全球研发的协调同步。

2021~2025 年医药创新生态系统中,“临床研究”和“监管审批”是两大贯穿其中的主题(图9),临床研究高效性和注册监管科学性受到更多关注。前者体现在临床研究执行、临床研究能力、临床研究体系保障3 个方面,对于我国临床试验受试者招募入组效率、试验数据质量提出更进一步的要求;后者体现在监管政策、监管标准和程序、监管体系3 个方面,在保障科学性的前提下充分利用全球临床数据在我国进行药品注册,确保在我国进行的国际多中心临床试验有充裕的招募时间窗口,且在试验数据锁定后可实现同步注册申请递交。此外,监管能力提升、临床人才培养、数字平台建设也是实现同步研发、注册与审评的有力保障(图10)。

二、推动同步研发、注册和审评的总体建议
当前,我国医药创新正步入更高层次的发展阶段。新冠肺炎疫情下,我国疫苗能够获得多国认可,并有三款已经列入世界卫生组织紧急使用清单,不仅展现了我国医药创新能力,更重要的是作为国际通用技术标准的制定和实施的实际案例,为我国药品监管逐渐实现科学化、国际化、现代化积累了宝贵经验。进一步推动在我国的同步研发、注册与审评将促使创新药更早地惠及患者,助力我国参与全球同步研发,提升我国药物研发水平和能力,更好地融入全球创新。进而使我国在已经处于全球医药创新第二梯队的基础上更上一层楼,不断完善与国际接轨的监管体系,提高研发水平和能力,适应新药研发全球化趋势,厚积薄发实现走向医药强国。
推动同步研发、注册与审评是一个需要各方政府、企业和研究者紧密配合的复杂工程。例如,实现早期研发的同步,需要优化遗传资源申请要求和流程,并大幅提高监管和临床的能力,确保每个环节的时效性;对于选择走路径1 的项目,企业需要在早期把我国纳入全球研发和注册的范围。对于路径1(早期临床试验即加入全球同步,通过实现各个阶段的同步研发,保障同步注册上市)和路径2(加入中后期全球多中心临床试验,实现同步注册上市),均需要包括国内外监管机构、国内外医药创新行业和我国研究者在内的各方积极参与推动国际数据互认,进一步落地实施ICH 标准。由于各种原因无法实现前两种路径,需要开展我国的桥接试验和单独临床试验的情况(路径3),需要完善和监管部门的沟通,使申办方提前规划。另外,行业各方可与监管机构通过早期沟通,积极探索其他可行路径,提升我国加入全球同步研发、注册与审评的比例。
本文重点提出10 条建议推动同步研发、注册与审评,包括解决当前瓶颈的主要举措、确保体系完善的主要抓手和推动持续升级的能力保障。
(一)解决当前瓶颈的“三大举措”
1. 提高遗传资源申请要求的合理性并优化流程效率
建立完善的、基于科学的、合理管控风险的人类遗传资源管理体系,建立有效的多方(跨部委、产业)沟通与对话机制,提高监管政策的透明度和审批时限的可预测性,针对性出台《人类遗传资源管理条例》实施细则。
2. 提升我国受试入组要求的科学性并增强国际数据互认
通过更加科学准确的系统方法,从临床研发项目招募实际难度、跨人种差异科学依据及统计学依据出发,决定我国受试者入组要求。在充分科学可靠的前提下,增加对东北亚临床试验数据的开放程度。
3. 推动临床机构流程的统一规范和协同并确保高效执行
理顺临床研究机构内的立项、伦理审批和合同签署的流程,通过统筹简化缩短临床试验启动时间;临床研究机构之间的流程统一可以通过卫生健康委等部门推进流程的标准化,并进行质量管控,将关键临床研究执行指标纳入机构评估。
优化伦理审批流程(如进一步落实推进伦理审批和临床申请的同步、统一各医院伦理委员会审批所需资料与流程),推进区域伦理委员会的建设与临床试验机构对区域伦理委员会的认可度,推动形成伦理审批质量的认证标准。
与此同时,申办方也应确保临床研究启动相关内部流程和沟通的高效。
(二)确保体系完善的“五大抓手”
1. 优化审评相关流程并鼓励以临床价值为导向的审评
优化加速审评申请流程(如可提前或在上市申请时提出优先审评申请),明确临床价值界定标准,鼓励原始创新的审评激励机制(如突破性治疗药物)。
2. 提高审评审批资料要求的合理性,进一步与国际接轨
进一步与国际接轨,减少不必要的特定文件的递交要求。
3. 推动与国际接轨的上市许可持有人制度全面落地执行
充分打通上市许可持有人跨境情形下的落地路径,保证集团化公司下属机构的上市许可持有人能力的充分协同(如药物警戒),明晰生产许可中对于分段、多场地、委托生产等的规定。
4. 推动临床研究机构平台打造和专职临床研究团队建设,促进机构明确定位,积累探索性临床试验的管理经验
打造专业临床研究平台,有效支持临床研究者并承载和发展相关学科。推动专职临床研究团队建设,打通职业发展路径并建立与工作内容相适应的考核体系。建议研究机构基于明确自身的定位,找准率先突破的领域和解决的问题,进而制定相应目标和计划,从而更加高效地不断积累探索性临床试验的管理经验。
5. 完善临床研究激励机制、资源投入
优化医院临床研究考核体系,提升临床研究重视程度,改革医生职称评定和绩效考核方法,鼓励医生开展和参与临床研究。保障临床研究投入,考虑增加临床医学研究在医学科学研究基金中所占比例,并推动设立临床专项科研计划或科研基金。
(三)推动持续升级的“两大保障”
1. 人才保障
加强监管队伍的建设,合理配置监管人员的数量,优化审评员待遇,丰富监管人才来源。构建研究、培训、实践相结合的教育培训体系,提高核心监管人才的质量,缩小不同地区监管能力的差距。加快建立跨境监管队伍,实现全球研发,全球化监管。
鼓励监管队伍秉持监管审评科学性的同时充分兼顾合理的灵活度。增强与发达市场监管机构的直接交流,更好地了解审评政策背后的原因和考量,确保监管执行中的因地制宜与持续现代化。
在学校教育中持续拓展临床人才相关课程的设置(如临床研究护士),并将临床研究人才的相关课程扩展到更多院校。全面推进职业教育,考虑在行业协会的牵头下,整合多方能力(如监管机构、高校、药企申办方、合同研究组织、临床试验全生命周期的第三方供应商),培养临床研究专才。
2. 体系保障
完善科学、透明、可预测的监管体系。逐步完成基于风险管理的理念转变及相应的管理能力的提升,优化管理效能;深度参与全球药品监管双边和多边合作机制,积极参加国际规范和标准的研究制定;大力推进药品监管科学研究,深化与高等院校、国家、地方以及民间研究机构的合作与协同。进一步统筹管理立法计划,加强对法规标准统筹管理,提高立法工作的透明度。
推进临床研究数字化工具平台建设,探索新技术在临床研究过程中应用。推进全生命周期数字化管理,推进监管和产业数字化升级。

来源:中国食品药品监管杂志


