您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-08-14 23:11
口服给药是最常用、最方便、最安全的给药途径,口服药品长期占据临床用药的主导和主体地位。然而,口服药物首先要被胃肠道吸收才能起效,这个过程非常复杂,会受到诸如药物理化性质,药物剂型类型(例如片剂,胶囊,液体制剂等)以及胃肠道生理环境等因素的影响。药物的理化性质加之人体的生理解剖特征,共同决定口服药物最终体内的暴露量以及在靶点位置的药物浓度,最终决定了药效的发挥。图1详细的描述了药物在体外给药后在人体内所要经受的层层屏障。
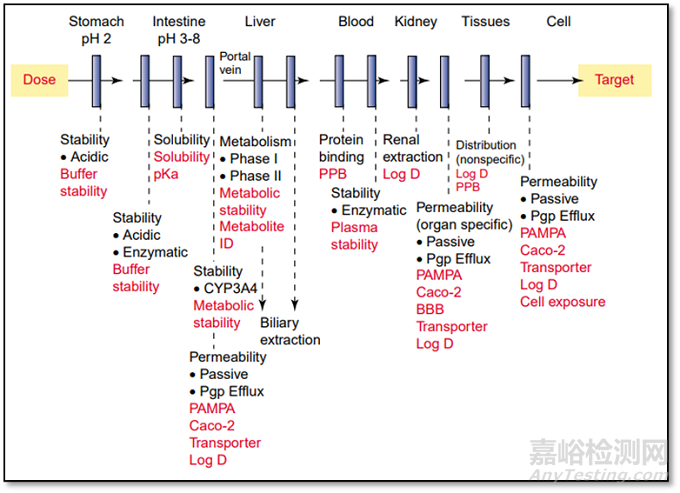
图1 药物体外给药后在到达靶点所需穿过的层层障碍
(来源于参考文献1)
药物口服给药后,化合物首先进入口腔。如果药物在口腔停留,那么可能被口腔粘膜的毛细血管吸收,这样也给口腔和舌下给药提供了依据。当然,药物制剂更多经过食道进入胃肠道。本文我们想详细谈谈药物吸收的第一站,也是药物吸收的第一道关卡——胃。
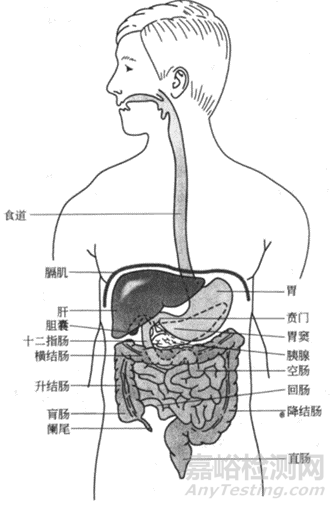
图2 人体胃肠道(来源于参考文献2)
1胃部解剖特点
人体的消化系统是由口腔到肛门的消化道构成,如图2所示,主要承担分泌、消化和吸收的作用。而胃是消化系统最膨大的部位,开口处称之为贲门,出口处称之为幽门,两者之间包括胃体部和前庭部。胃壁由外向内分为三层:粘膜-肌层-浆膜。粘膜收缩可见褶皱,在其表面有无数的凹陷,但是胃部没有绒毛,故而其吸收表面积要小的多,因此胃对于药物和营养物质吸收较差(胃表面积较小,大约1平方米),当然也和药物在胃部停留时间较短以及胃周围血流量较少有关。
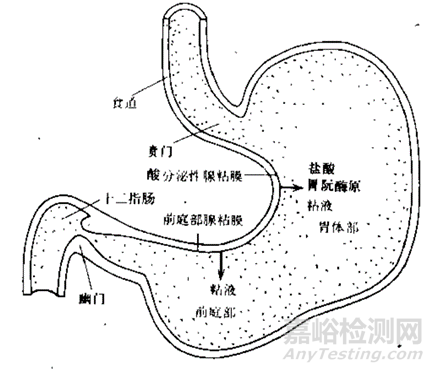
图3 人体胃部构造(来源于参考文献3)
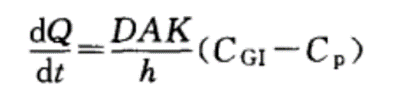
被动扩散是大多数药物吸收的主要方式,而被动扩散是药物自发地由高浓度一侧向低浓度一侧扩散,无需消耗能量,可以用Fick’s方程来描述。Fick’s方程中dQ/dt是扩散速度,D是扩散系数,A膜表面积,K是药物的分配系数,h是膜厚度,CGI为胃肠道药物浓度,Cp为血浆中药物浓度。由于血浆中药物快速的运送到全身,浓度可以忽略不计。从上式可知,药物吸收与药物的亲脂性,吸收膜的厚度,表面积以及药物的溶解度有关。其实结合Fick’s方程,也就解释了为什么胃部吸收表面积要小,停留时间较短以及胃周围血流量较少,药物更多的在小肠吸收的原因。
2药物在胃中理化性质变化
人体口腹服用250ml水后,胃中的pH为1.2-7.4人胃中的余留液体的体积约为30ml。餐后状态下,胃中的pH可增高至7,并在1.5h后逐步恢复至正常水平。
众所周知,大多数药物为弱酸或者弱碱性药物,胃肠道pH的变化,会影响酸碱性药物的解离型和未解离型的比例,进而影响药物的溶解性和渗透性,如图3所示,对于一个pKa=5的弱酸性药物,当pH>5时,即pH>pKa,药物离子型形式的比例大于其非离子型形式,离子型形式的增多有助于药物与水性介质发生静电相互作用,静电相互作用一般要强于其他非共价相互作用力(常见为氢键作用力),进而增加药物的溶解度;但是离子型的增多,极性的增加,降低了其logP值,即亲脂性降低。药物要想吸收入血,需要跨膜,细胞膜在内的生物膜为磷脂双分子层,关于亲脂性,通常非解离型(分子形式)化合物的脂溶性高于离子型。对于此酸性药物,渗透性降低。当pH<5时,即pH<pKa,对于此酸性药物,更多的分子形式存在,溶解度降低,渗透性增加。
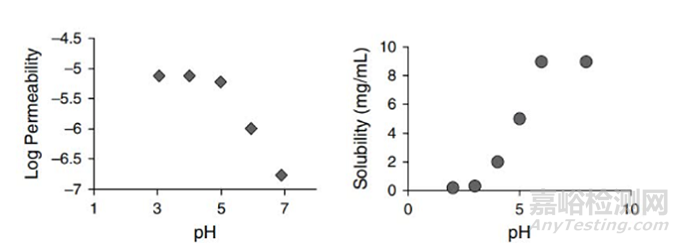
图3 pKa=5的酸性药物溶解性和渗透性随着pH变化而发生的变化
(来源于参考文献4)
众所周知,药物只有溶解才能吸收,药物吸收以后才能发挥作用。对于一个药物来说,其理化性质是不同的,这样就会在胃肠道处于不同的状态,进而影响其吸收的情况。
一般来说,酸性药物在胃部的酸性环境更多的以分子形式存在,可能存在吸收。而对于碱性药物在胃中几乎是不存在吸收的。当然,还要结合药物的理化性质,胃肠道的解剖特征以及胃肠道的生理状况,具体情况具体分析。
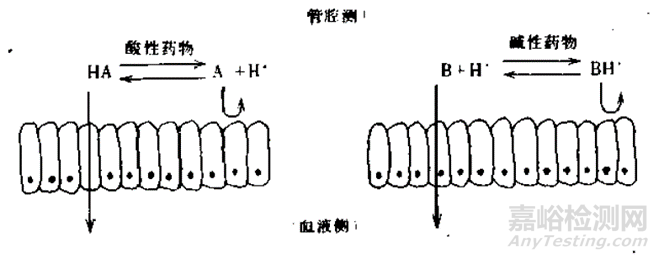
图4 酸碱性药物在胃肠道的解离与透膜行为(来源于参考文献3)
3胃排空与药物吸收
胃是一个囊状器官,表面相对平滑。胃能吸收物质,但对于整个胃肠道吸收来说胃的贡献是有限的。近端小肠的吸收优于胃和肠道其他任何部位。药物从胃进人小肠的速度是吸收的一个限速步骤。食物由胃进入十二指肠的过程称之为胃排空。
在“普通口服固体制剂溶出度试验技术指导原则”中提到“在禁食状态下,胃内滞留(排空)T50%时间为15∼20分钟。对于高溶解性-高渗透性(1类)及某些情况下的高溶解性-低渗透性(3类)药物制剂,以0.1mol/L HCl为介质,在适当的溶出度试验条件下,15 分钟的溶出度大于85%时,可认为药物制剂的生物利用度不受溶出行为的限制,即制剂的行为与溶液相似。在这种情况下,胃排空速度是药物吸收的限速步骤。如果药物制剂溶出比胃排空时间慢,建议在多种介质中测定溶出曲线。”,可见,胃排空的时间影响药物制剂(尤其是速释胶囊和片剂)在胃中的滞留时间,进而影响药物的吸收。
胃的运动很复杂,受神经和激素调节。胃排空速率与节率性收缩有关,禁食时,大约每分钟收缩3次,当食物进人胃时收缩频率更小。进食后,胃排空速率常常与胃内容物的形态(固体或液体)和营养组成有关:固体食物一般在进食20-40 min后开始进行胃排空,而液体的排空则没有延迟,并且随着液体营养密度的增大,胃排空逐步减缓。摄入膳食纤维、脂肪、蛋白质和低升糖指数食物亦可减缓胃排空。固体食物的胃排空与热量有关,两者呈现反比的关系,即热量越高,排空越慢。任何药物或食物只有在胃消化成适宜的程度,才能够进人近端小肠,这个过程是一种保护机制,能避免小肠精巧的吸收表面受到损伤。液体食物能促进胃排空,固体则延缓排空。促进作用是由于激活胃壁上的牵张感受器而产生的。当流体是水时,抑制性感受器激活停止,加速胃内容物的排空。
4总结
总之,上市药物中口服药物占据大多数,而对于口服药物来说,多经过胃肠道进行吸收。胃作为口服固体制剂崩解成小颗粒,小颗粒解聚,继而溶出的主要发生场所。胃可能更多发挥消化的功能,但是胃的解剖学特征及其生理状况,特别是胃排空,对于药物的吸收发挥着举足轻重的作用。药物制剂的开发涉及多学科的知识与理论,努力去健全生物药剂学以及解剖生理在制剂开发中的作用,知道此种药物制剂的“来处”,明了其“去处”,才能更好的做出更加符合安全性,稳定性,有效性及可生产性的制剂产品。
参考文献
1.Pharmaceutical profiling in drug discovery
2.应用生物药剂学与药物动力学(图书)
3.图解药剂学(图书)
4. Drug-like Properties Concepts Structure Design and Methods; from ADME to Toxicity(图书)
5.食物影响口服药物吸收的研究进展
6.胃排空速率变异与血糖调节的关系及基于胃排空速率的降糖策略
7.制剂百科全书(图书)

来源:药渡


