您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-08-15 13:52
固体制剂是最常用的给药方式,药物以片剂或胶囊给药后历经以下四个阶段进入血液循环,发挥作用:①药物由胃排空和肠道的转运达到吸收部位;②药物在胃或者肠道溶出;③溶解的药物经过胃肠道生物膜进入循环系统;④吸收后的药物经肝脏(首过效应)并到达体循环。我们可以简单地把口服固体制剂的体内过程分为四个阶段,当然,不同的药物制剂这四个阶段呈现的顺序略有差别。

药物口服生物利用度是从数理的角度去描述药物从吸收到入血的过程。药物的口服生物利用度(F)是指机体从药品中吸收API或活性物质并在作用部位产生药效的程度和速度,是药物吸收分数(Fa)、药物排出肠道代谢的分数(Fg)和药物排出肝脏代谢的分数(Fh)的乘积,如方程1所示,Fa为吸收分数,Fg为胃肠道代谢后的剩余分数,Fh为肝脏首过效应后剩余的分数。
药代动力学是描述药物的吸收、分布、代谢和排泄(ADME)过程的科学。药物的吸收是指药物从给药达到体内循环的速度与程度。药物的吸收情况决定了药物达到靶标的时间与程度。药物通过以下转运方式吸收:被动扩散,载体介导的主动转运,细胞旁路以及细胞内吞作用。被动扩散是大多数药物吸收的主要方式,而被动扩散是药物自发的由高浓度一侧向低浓度一侧扩散,无需消耗能量,可以用Fick’s方程来描述。

Fick’s方程中dQ/dt是扩散速度,D是扩散系数,A膜表面积,K是药物的分配系数,h是膜厚度,CGI为胃肠道药物浓度,Cp为血浆中药物浓度。
1小肠解剖学

图1 小肠示意图(来源于参考文献1)
药物吸收主要发生在小肠部位。小肠由十二指肠,空肠和回肠组成,解剖长度可到680cm,生理上活动长度约2-3m(282cm),直径约4cm。十二指肠与胃相连,胆管与胰腺开口于此,排出胆汁和胰液,帮助消化和中和部分胃酸使消化液pH升高。

图2 十二指肠示意图(来源于参考文献1)
1十二指肠
十二指肠生理活动上的长度约为21cm。十二指肠内具有胰腺和胆囊管的开口,由于肠中的碳酸氢盐中和胃中排出的酸性食糜,使十二指肠的pH升高,十二指肠中的pH为4-6,也说6-6.5。这是酶消化蛋白和肽类食物最适宜的pH环境,富含各种消化酶的胰液通过胆汁管分泌到十二指肠中,可用于消化蛋白,碳水化合物和脂肪,这样就构成了复杂了环境,有助于难溶性药物的增溶。
2空肠
空肠生理活动上的长度约为105cm。空肠为小肠中段,位于十二指肠与回肠之间。蛋白质和碳水化合物接着被胰液进行消化。空肠段收缩频率较低,有利于进行药物吸收的体内研究。
3回肠
回肠生理活动上的长度约为156cm。回肠为小肠末端,pH约为7,远端可达到8。因为碳酸氢盐的存在,酸性药物易于溶解;分泌的胆汁可以增溶脂溶性药物。
2小肠生理学与药物吸收
1小肠巨大的表面积
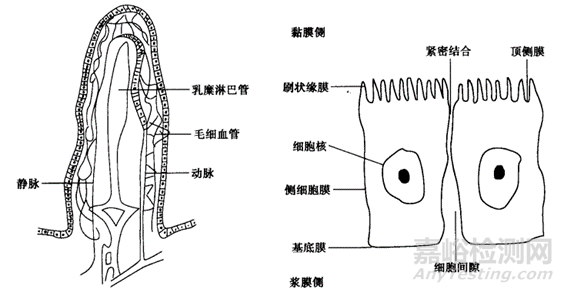
图3 左侧为绒毛,右侧为微绒毛示意图(来源于参考文献2)
小肠黏膜面上分布有许多环状褶襞,并拥有大量指状突起的绒毛。绒毛是小肠黏膜表面的基本组成部分,长度约0.5~1.5mm,绒毛内含丰富的血管、毛细血管以及乳糜淋巴管,是物质吸收的部位。每一根绒毛的外面是一层柱状上皮细胞,其顶端细胞膜的突起称为微绒毛。每一柱状上皮细胞的顶端约有1700条微绒毛,是药物吸收过程进行的区域。微绒毛上的细胞膜约厚l0nm,上皮细胞面向黏膜侧的膜称为顶侧膜,构成刷状缘膜。面向浆膜(或血液)侧的膜称为基底膜,细胞两侧膜称为侧细胞膜。相邻细胞之间充满间隙液,其细胞顶侧膜处相连,构成紧密结合,这是细胞旁路通道的转运屏障。

图4 小肠解剖图示从左到右如何增加药物吸收表面积(来源于参考文献4)
图4详尽地描述了小肠的解剖学结构如何具有如此大的表面积。在图的左侧,肠道的吸收表面积被描绘为标称值为1的圆柱体。在第二张图中,表示了粘膜皱襞。它使得肠道的表面积增加了 3倍。下一张图描绘了从肠黏膜表面延伸到整个肠长的指状绒毛。绒毛的尺寸因每个物种而异,取决于每平方毫米粘膜表面的绒毛的长度、宽度和密度。绒毛使吸收表面增加5-10倍。最后一张图说明了单个肠细胞的表面。其中的刷状缘由微绒毛组成,可将吸收表面积增加20-25倍。
2小肠转运时间
由于小肠部位环状褶皱,绒毛,微绒毛的存在,使小肠具有广泛的与药物接触的表面积,约达200平方米;从表1可知,药物在小肠相对较长的滞留时间,由Fick’s方程也可推出小肠为药物的主要吸收部位。小肠转运时间SITT为3.5h,与胃排空相比,受制剂的尺寸大小影响不大,然而与自身调节波有关,饭前45min服用片剂可以将现场转运时间显著降低至1.7-2.4h。
表1 胃肠道生理与药物吸收(来源于参考文献2)

3胆汁胶束
胆盐类(如牛黄胆酸盐,甘胺胆酸盐)在胃排空状态下由胆囊释放进入十二指肠,形成胶束。胆汁胶束不仅可以增溶,还会降低游离药物的百分数,进而降低药物的渗透性。胆汁胶束的浓度具有很大的物种差异和餐后/空腹差异。由于羧酸根或者硫酸根的存在,胆汁胶束带有负电荷。因此药物的亲脂性以及其所带电荷都会影响药物与胆汁胶束的作用。胆汁胶束与表面活性剂有所不同,其不具有临界胶束浓度。
4肠中代谢
肠上皮细胞中主要的代谢酶细胞色素P450 3A4同工酶。药物可在肠中被代谢,肠代谢为首过效应的一部门,为药物的系前代谢。
5肠中水解
肠道中的丰富的消化酶也可以水解药物,特别是酯类,酰胺类及氨甲酸酯类药物酶存在于肠腔,特别是刷状缘浓度较高。所以研究化合物在模拟胃肠道环境中的溶液稳定性,全面表征对化合物的体内实验做到心中有数。
6胃肠道灌流
血液灌流对于胃肠道药物吸收起到举足轻重的作用。十二指肠及腹膜都有丰富的毛细血管和淋巴管网灌流。药物经小肠吸收后通过肠系膜血管经肝门静脉进入肝,然后进入全身循环。药物也可经微绒毛下的乳糜管或淋巴管进行吸收,这部分药物不经过肝门静脉,可以避免肝脏的首过效应。
3总结
口服制剂以其得天独厚的优势仍旧为药物剂型的主流。无论是简单的描述药物从口服到吸收进入体循环,还是生物利用度的角度抑或ADME角度去剖析药物分子理化性质及制剂特征与生理解剖学发生的“化合反应”,都需要制剂开发人员生物药剂学的知识具有全面的认识。
当然,对于一个已经设计好的候选化合物,制剂人能做的更多的以制剂手段改变其吸收的速度与程度,也就是可以控制制剂的溶出,进而影响药物的吸收。至于药物的分布,代谢(包括系前代谢)及排泄则与药物分子的特性相关,药物从制剂溶出以后,制剂人很难再去控制。希望以上对于生物药剂学的理解,能有助于帮助大家解释药物在体内的变化,从而开发选择更佳有效药物分子。
本文详细的介绍了消化系统中对于药物吸收最重要的部位-小肠。研究的项目及候选化合物的性质是不断变化的,但是胃肠道环境所具有的一般规律是不变,熟悉胃肠道的特征,才能以不变应万变。希望这篇文章能够给大家一些帮助。
参考文献
1. 人体解剖生理学 K.M.范德赫拉夫(书籍)
2. 生物药剂学与药物动力学 主编:梁文权(书籍)
3. Drug-like Properties Concepts Structure Design and Methods; from ADME to Toxicity(图书)
4. Contrasting the Gastrointestinal Tracts of Mammals: Factors that Influence Absorption

来源:药渡


