您当前的位置:检测资讯 > 监管召回
嘉峪检测网 2022-10-22 23:07
近年来,应用替代终点来进行药物评价和监管决策已经成为提高药物研发效率的重要手段。美国和欧盟已经建立了较完善的基于替代终点的新药上市审批程序,替代终点在我国的新药上市审批中也发挥越来越重要的作用。本文从替代终点在新药上市审批中应用的原理入手,对美国和欧盟基于替代终点的新药上市审批程序进行深入研究,并对我国基于替代终点的新药上市审批程序的完善提出建议。
2020年7月1日,我国正式实施了新的《药品注册管理办法》,确定了4条新药快速上市路径。其中,附条件批准路径创新性地将替代终点应用到新药上市审批中。早在1992年,美国通过修订《联邦行政规则》创设了加速审评( accelerated approval, AA) 途径,制定了基于替代终点的上市批准程序,并在2012年颁布的《食品药品安全与创新法 案》 ( Food and Drug Administration Safety and Innovation Act,FDASIA) 中将这一程序从法律层面确认下来。欧盟也于2005年开设了附条件批准( conditional mar- keting authorization,CMA) 上市路径,允许基于替代终点给予特定新药有条件的批准,以加速临床研发。近年来,应用替代终点进行药物评价和监管决策已经成为提高药物研发效率的重要手段。本文将从替代终点在新药上市审批中应用的原理入手,结合美国和欧盟应用替代终点进行新药审批的经验,为我国替代终点在新药上市审批程序中的应用提供建议。
1 替代终点在新药上市审批中的应用原理分析
1.1 替代终点的概念与实质
临床试验是评估一种新药安全性和有效性的科学标准方法,而临床终点则被认为是临床试验中评估治疗干预效果的金标准。但在有些情况下,临床终点有时难以获得,或者需要经过大规模、长时间并且耗资巨大的临床试验研究,这不仅延误了新药的审批进程,还会加重制药企业的研发负担[1]。替代终点( surrogate endpoints,SEPs) 是指用于间接反映临床结局( clinical outcome) 的终点指标,包括实验室检查项目、放射影像学、体征或其他指标,可用于替代临床终点或预测临床获益及损害。相比较于临床终点,替代终点可以更早、更方便地测量,因此将替代终点应用于新药审批中,可以更快地促进新药上市、节约医药企业的研发成本。
替代终点的实质是一种基于流行病学、治疗学、病理生理学或其他科学依据的生物标志物( biomar- kers,BMs) [2]。生物标志物是能够被客观测量和评价,反映生理或病理过程,以及对暴露或治疗干预措施产生生物学效应的指标[2]。根据生物标志物的用途和它们提供的信息类型对其进行分类,可以分为靶向生物标志物、机制生物标志物、结果生物标志物、毒性生物标志物、药物基因组学生物标志物和诊断性生物标志物。其中结果生物标志物是与疾病或相关病理状况有明确关系的生物标志物,可作为评价候选化合物效果的指标,并应用于新药审批中。但是,并非所有的生物标志物都可以作为疗效验证的替代物,其有效性需要科学规范的研究验证。经过充分验证后,还需要经过监管机构审评或认定为足够有效、可以作为检验临床试验疗效的指标,生物标志物才能被称为替代终点[3]。因此,生物标志物成为替代终点,有2个非常重要的程序: 验证( vali-dation) 和认定( qualification) 。
1.2 替代终点的验证
替代终点的验证指的是通过稳健的统计方法确认候选替代终点满足一系列应用于临床的充分和必要条件[4]。替代终点本身并不直接反映临床结局,只是对临床结果起到预测作用。生物标志物成为替代终点,必须有明确和令人信服的科学证据验证其能够持续和准确地预测临床结果,否则不能成为替代终点。学术界存在很多替代终点的验证标准,最严格的标准是Prentice提出的,主要归结为两点: ① 作为替代终点的生物标志物必须与真实的临床结局相关。② 作为替代终点的生物标志物必须完全反映干预对临床结局的净影响[5]。只有2个条件都满足的情况下,才能认为生物标志物是合格的替代终点。
1.2.1 验证原理 在早期的临床试验中,对替代终点的应用存在误区,认为只要是和临床结局相关的生物标志物便可以用作替代终点[6]。多起失败的案例为替代终点的应用敲响了警钟。Fleming等[7]指出,相关性是不足以构成生物标志物成为替代终点的证据的,它们之间必须具有因果关系,即干预措施对替代终点的任何改变都将转化为临床结局的相应改变,且替代终点处于疾病到临床结局的唯一因果通路上( 图1E) 。然而由于疾病机制的复杂性,往往会出现替代终点不在疾病到临床结局的因果通路上( 图 1A) ,或者疾病到临床结局的因果通路并非唯一的,替代终点不能完全反映干预对临床结局的净影响( 图 1B、图 1C、图 1D) 。
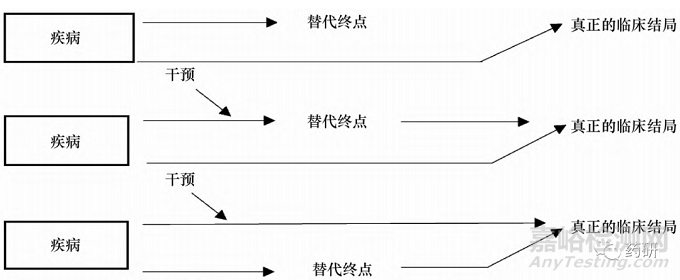

为了降低不可靠的替代终点对临床结局的预测产生的误导信息所带来的风险,必须对替代终点进行验证。支持验证的科学证据则需要通过开展广泛的临床试验来获取[8]。
1.2.2 验证流程 替代终点的验证工作一般由企业或研发机构完成,并向监管机构提交认定申请。同时,替代终点的开发和验证通常内嵌于药物的研发过程中,从临床前研究就开始。目前关于替代终点的验证还未形成统一的标准,但是验证基本流程可以总结如下。
首先基于病理生理学研究和对疾病过程的理解,对生物标志物与临床结果之间关联的生物学合理性进行确证,具有生物学合理性是生物标志物能够作为候选替代终点的最低证据标准; 接着通过流行病学研究和观察性研究,进一步探究生物标志物与临床结果之间的统计学相关性; 再进行随机对照临床试验,在充分和良好控制的临床试验中,根据候选替代终点的变化,利用数学统计方法推导出临床结果的预期效果。同时,验证过程应当考虑可能出现候选替代终点的预测与临床结果无关的情况,建立暴露( 治疗干预) -反应模型来描述和预测药物剂量或血浆浓度,以明确替代终点与临床结果之间的关联的真实性[9]; 最后,对多个临床试验进行Meta分析,进一步确定采用不同的药物类别和在疾病的不同阶段实施干预措施后,其对替代终点造成的任何改变都将转换为临床结果的相应改变,即替代终点与临床结局之间存在真实的因果关系[7]。随着验证流程的逐步推进,替代终点验证的证据强度逐步增强、候选替代终点的数量逐步减少,同时纳入试验研究的病例数也逐步增加。但是在实际的验证过程中,受限于临床试验开展的诸多条件的限制,往往并非都能获得最高等级的证据标准。见图 2。

1.3 替代终点的资格认定
替代终点被药品监管部门采纳并应用于药品审评决策中,有2种途径: 一种是将替代终点开发和药物研发捆绑。在药物研发的过程中,研发机构将替代终点的开发和验证数据等提交给药品监管部门,药品监管部门基于对数据的技术评判与研发机构开展沟通交流,决定是否运用此替代终点支持审评决策。这一途径的优点是可以提高药物的研发效率,缺点是这一途径下替代终点的数据不能被其他研发项目共享,也不能运用于其他药物审评决策中[10]。另一个途径就是通过资格认定程序,成为共享的药物研发工具。FDA将资格认定界定为: 对某医疗产品研发工具是否可以在特定的应用场景下( context of use,COU) 应用于药品的开发和监管审查的确认[11]。特定的应用场景指的是医疗产品研发工具的使用方式和用途。一旦通过资格认定,资格认定的相关信息就可以公开获取,并可以应用于其他药物研发项目中。2010年,美国FDA的药物评价和研发中心( Center for Drug Evaluation and Research, CDER) 就制定了生物标志物认定的资格指南,并于2015年建立了“药物研发工具资格认定程序”( qual- ification process for drug development tools,QPDDT) ,其中最重要的一项内容就是生物标记物资格认定项目 ( biomarkers qualification program,BQP ) 。2016年,美国颁布了《21 世纪治愈法案》( 21st Century Cures Act) ,并将药物研发工具资格认定程序正式纳入了这一法案中。2014年11月10日,欧洲EMA发布《药物开发新方法的资格鉴定: 申请人指南》,该指南为申请人开发生物标志物等新方法提供了运营、费用、程序等方面的指导。欧洲议会和理事会( European Parliament and of the Council) 的第 726 / 2004号法规为在药品开发框架内提供生物标志物使用的科学建议提供了法律基础[12]。
1.3.1 认定流程 美国和欧盟在生物标志物的认定流程上基本上是一致的,包括启动、咨询和建议、审查3个阶段。为了提高资格认定的效率,美国FDA和欧洲EMA鼓励申请人向2个机构同时提交平行资格认定申请[13]。美国和欧盟的生物标志物资格认定流程和沟通框架见表 1。



1.3.2 认定层级 美国FDA和欧洲EMA对替代终点的认定层级主要基于生物标志物作为替代终点进行验证时所获得的证据等级。根据证据等级的不同,将申请认定的替代终点分为不同的层级。美国FDA与欧洲EMA的替代终点的层级划分大体一致,主要是根据ICH-9指南和美国国立卫生研究院( National Institutes of Health,NIH) 生物标记物定义工作组框架发展而来的,分为3个层级: 已验证的替代终点、可能有效的替代终点和候选的替代终点,见表 2。
2替代终点在美国和欧盟新药上市审批程序中的应用
替代终点的认定层级不同,在新药上市审批中的应用也有所区别。已验证的替代终点经过了随机对照临床试验的验证,与真正的临床终点几乎具有同等的证据水平,可以替代临床终点。因此,在符合风险效益比的前提下,可以直接应用于常规审批( ordinary review) ; 可能有效的替代终点的验证证据基于统计推理和临床观察的集合,可以合理地预测临床结局,但是不能完全替代临床终点。因此,这一层级的替代终点不能用于常规审评。但是基于风险效益或是公共卫生方面的考虑,美国FDA和欧洲EMA允许将其用在严重或危及生命的疾病药品的加速审评程序中,如美国FDA的加速审评程序和欧洲EMA的附条件批准程序中,候选替代终点则因为验证的证据级别较低,还需要后续的持续评价,因此不能作为药品审评的依据。但是这类替代终点在缺乏更加可靠的临床实践证据的前提下,可以有助于疾病诊断、预后评估或者由治疗人员指导患者进行疾病病程管理[2]。

2.1 已验证的替代终点在常规审批中的应用
经过常规审批路径上市的新药需进行基础研究、临床前研究,随后通过Ⅰ~ Ⅲ期临床试验,直至达到真正的反映临床获益的临床终点才可获得批准上市。经过验证的替代终点虽然不能直接反映临床获益,但是可以可靠地预测临床获益,因此可以通过常规审批路径被批准上市。由于替代终点可以更早期地被监测到,因此会大大缩短临床试验时间,加快药品上市的进程,降低药物研发的总成本。同时,那些未能影响替代终点的候选化合物的临床试验可以比旨在监测临床终点的研究提前结束。临床试验的早期终止可以减少患者对失败候选化合物的暴露时间,从而降低患者的风险[14]。
能够应用于常规审批的替代终点通常需要满足以下几个标准: ① 明确界定替代终点应用的患者人群、治疗的疾病或用途以及替代的临床终点。② 替代终点和临床结局之间具有强烈的统计学相关性。③ 替代终点必须在至少 1 项临床试验的每例患者个体资料( individual patient data,IPD) 中显示与临床终点具有明确的关联。④ 以上关联须在多个临床试验数据中获得验证[15]。由于通过验证的替代终点所需的证据级别较高,因此比较难以获得。但是一旦通过验证和认定,则可以按照常规审批流程被批准上市。批准上市后,按照常规开展上市后研究即可。针对常规审批,美国 FDA 和欧洲 EMA 分别设立了加快上市审批程序: 包括美国 FDA 的快速通道 ( fast track) 、突破性疗法认定 ( breakthrough therapy) 和优先审评( priority review) 程序,以及欧洲EMA 的加速审评( accelerated assessment) 程序。如果同时满足这些加快上市审批程序所需的条件,则可以同时被纳入其他加快上市审批程序。
2.2 可能有效的替代终点在加速审批中的应用
可能有效的替代终点应用于上市审批程序中至少需要满足以下几个标准: ① 明确界定替代终点应用的患者人群、治疗的疾病或用途以及替代的临床终点。② 替代终点和临床结局之间具有强烈的统计学相关性。③ 有大量的临床证据表明替代终点测量的临床结局能够准确预测真正的临床结局。④ 当干预措施是安全的,有大量的临床证据表明替代终点偏离临床终点的风险在可接受范围内[16]。同时,美国FDA和欧洲EMA规定,被批准基于可能有效的替代终点提起的上市申请可同时被纳入FDA的优先审评程序和EMA的加速审评程序。
由于可能有效的替代终点没有经过充分的验证,因此应用于新药审批中会存在以下风险: ① 使用可能有效的替代终点会提前结束上市前的临床试验,使得很多罕见或迟发性不良事件导致的药品风险被忽略。② 替代终点在成本-效益评估方面的可靠性是有限的,使用替代终点可能阻碍对具有真正临床终点的大型、实用的临床试验的投资。③ 对于非盲法的观察性研究而言,应用替代终点会使已验证的临床终点的适用范围扩大,不确定性风险增加。如NIH发起了一项随机、安慰剂对照的心律失常抑制试验,以确定莫雷西嗪、氟卡尼或恩卡尼抑制室性早搏收缩( premature ventricular contractions,PVCs)是否会降低心肌梗死的死亡率。试验结果显示该药对替代终点PVCs非常敏感,但这种“有益的结果”却导致心脏骤停或心律失常相关死亡率上升了364%[17]。
2.2.1 可能有效的替代终点和已验证的替代终点审批程序比较 美国 FDA和欧洲EMA都针对可能有效的替代终点设置了特殊的审批程序,即FDA 的AA审批路径和EMA的CMA审批路径,并通过相应的配套措施来降低风险。与应用已验证的替代终点的常规审批程序相比,应用可能有效的替代终点的药品审批程序具有以下几个特点。
① 限定适用范围。监管机构药品审批决策的最重要的指标就是风险-效益( risk-benefit) 比。在大多数情况下,药品的效益必须明显超过风险才能被认为适合商业使用。因此,为了使得风险-效益比可接受,基于可能有效的替代终点申请上市的药品需要具有更高的效益。因此,主要解决未满足的医疗需求、公共卫生紧急情况、治疗罕见疾病等具有更高社会效益的药品等才能基于可能有效的替代终点申请上市。
② 提起意向申请。监管机构对基于可能有效的替代终点的上市申请做出审评决策时,需要额外考量不同于常规审批的一些证据标准,需要早期介入和申请人开展充分的沟通交流,需要考察申请的药品是否落入了限定的适用范围,还需要审核申请人的证据补充计划等,因此需要在提交上市申请之前向药品监管机构提交意向申请。
③ 额外的证据标准。除了提交常规审批所需的基本资料以外,拟申请应用可能有效的替代终点上市的药品申请还需要提供额外的证据,证明其基于可能有效的替代终点被批准上市的合理性和可行性。
④ 早期沟通交流介入。为了提高审评决策的质量和效率,降低决策风险以及促进新药上市,药品监管部门会在药品研发的关键阶段和申请人开展沟通交流。对于提起基于可能有效的替代终点上市的意向申请,美国FDA和欧洲 EMA都会在其提交上市申请之前就关键技术性问题和申请人开展充分的沟通交流,探讨可否应用替代终点指标,以及验证性临床试验的有关问题,并提供一定的科学建议。
⑤ 上市后确证性临床研究和再评价。基于可能有效的替代终点申请上市的药品的临床试验一般受试者规模偏小或持续时间偏短,确证性的临床试验证据不全,临床获益具有不确定性[18]。因此要求申请人经批准上市后继续开展上市后的确证性临床研究以证明其临床获益,并收集各种安全性数据。同时美国FDA和欧洲EMA基于上市后研究证据对药品开展再评价,决定对上市许可进行维持、变更、暂停或撤销。
⑥ 分批授权程序。经过常规审批程序上市的药品取得的是药品监管机构颁发的完全上市许可( full approval) ,开展常规的上市后研究即可。美国FDA 基于上市后研究结果和药品安全风险监测情况,决定药品是否应继续存留在市场或采取何种监管措施[19]。欧洲EMA要求常规程序上市的药品定期提交上市后的安全更新报告、积极开展药品不良反应监测,并通过药品再注册制度对上市药品的风险-效益进行重新评估,建立药品的市场退出机制。
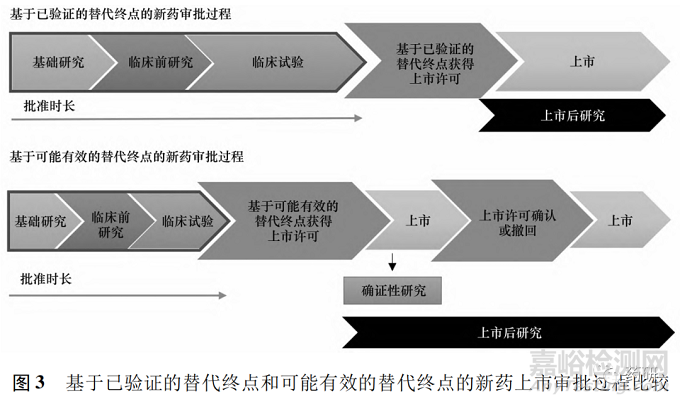
基于可能有效的替代终点上市的药品拿到的上市许可属于附条件性的许可,获得批准后需要继续开展确证性研究,证明药品的风险效益比在可接受的范围,否则上市批准将被撤销。同时,如果完成了上市后的确证性研究,并且获得支持药物效益大于风险的完全证据,则可以将许可转换为完全上市许可。基于已验证的替代终点和可能有效的替代终点的新药上市审批过程比较见图 3。
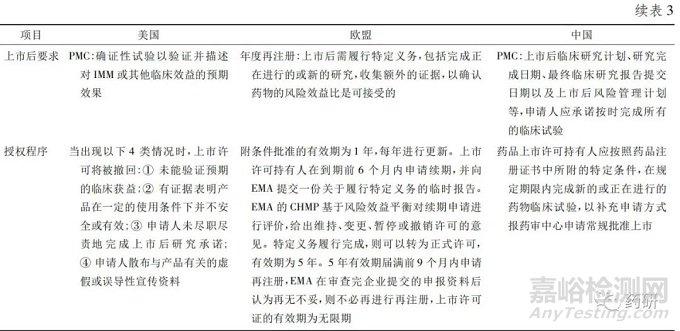
2.2.2 美国FDA和欧洲EMA基于可能有效的替代终点的新药审批程序的应用 美国FDA基于可能有效的替代终点的上市审批程序为AA,欧洲EMA 则为CMA。表3 对这2个程序从法律依据、适用范围、额外证据、沟通交流、市后要求和授权程序等方面进行论述和比较。


3替代终点在我国新药上市审批程序中的应用及启示
我国在2017年发布的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》中首次明确提出: 对治疗严重危及生命且尚无有效治疗手段疾病以及公共卫生方面等急需的药品医疗器械,临床试验早期、中期指标显示疗效并可预测其临床价值的,可附带条件批准上市。2019年修订发布的《药品管理法》提出: 对治疗严重危及生命且尚无有效治疗手段的疾病以及公共卫生方面急需的药品,药物临床试验已有数据显示疗效并能预测其临床价值的,可以附条件批准,并在药品注册证书中载明相关事项。2020年3月,新的《药品注册管理办法》发布,进一步确认了我国建立附条件批准加快上市路径。随后,国家药品监督管理局于2020年7月发布了《药品附条件批准上市申请审评审批工 作 程 序( 试 行) 》、2020年11月发布了《药品附条件批准上市技术指导原则( 试行) 》( 简 称《技术指导原则》) 。其中,《技术指导原则》里面明确了可以依据替代终点批准药品上市。因此,我国已经建立起了基于替代终点的药品加快上市审批路径。
3.1 我国基于已验证的替代终点的新药上市审批程序发展现状及启示
我国在《技术指导原则》里面明确提到依据已知能够合理预测临床获益的指标可用于常规批准。根据美国和欧盟的经验可知,将替代终点应用常规审批路径,关键在于对替代终点进行验证和认定。但是目前我国尚未出台一部关于替代终点验证和认定的指导原则,相关工作只是停留在国内科研机构的少数验证项目[3]。美国和欧盟基本上建立了较为完整的替代终点验证认定指南和制度,并且发布了经过认定的替代终点清单。基于列入清单内的已验证的替代终点的上市申请可以直接纳入常规审批路径。因此,对于基于已验证的替代终点的新药审批程序提出以下建议。
3.1.1 鼓励替代终点的开发 在鼓励替代终点的开发方面美国FDA提供了可借鉴经验: 一是需要监管部门出台相应的政策进行引导和支持。FDA通过采取发布支持信( letter of support,LOS) ,向研发机构和人员公布具有潜力的替代终点,并提出研发倡议,鼓励和刺激替代终点的研发。并通过关键路径创新会议( critical path innovation meeting,CPIM) ,与研究人员探讨替代终点开发相关的创新方法和技术,并提供专业指导,促进替代终点的开发[20]。二是构建政府、企业、学界、基金会等多方参与的替代终点研发平台,引入公私合作伙 伴( public-private partnership,PPP) 模式,形成智力资源、人力资源和资金的聚集,加快替代终点的研发进程。2006年3月,美 国FDA倡导成立了药物安全性预测联盟Predictive Safety Testing Consortium,PSTC) ,合作成员包括19家大型国际制药公司以及FDA,EMA及日本药品与医疗器械管理局( Pharmaceuticals and Medical Devices Agency,PMDA) 等政府监管机构[3],为替代终点的开发提供了有效支持。
3.1.2 完善替代终点验证认定指南 美国FDA自2016年起,陆续发布了《生物标志物认定举证标准规定架构》、《针对行业及FDA人员生物标志物认定的举证架构指南》( 草案) 、《药物研发工具的通用流程指南》、《生物标志物证据框架指南》和《生物分析方法验证指南》等一系列替代终点认定验证指南。欧盟也建立了较为完善的验证认定制度和行业标准。尽快建立我国的替代终点验证认定指南是完善基于替代终点的药品审批路径的核心。
3.1.3 发布替代终点认定清单 替代终点的验证需要大量证据的累计,消耗大量时间和资源。建立替代终点认定清单制度,并且公开替代终点验证数据,有利于数据共享和额外数据的积累。FDA的替代终点清单针对成人和儿科患者分别列举,内容包括疾病/用途、患者人群、替代终点、适用于的批准类型( 常规审批或加速审批) 、作用机制、适用年龄范围( 儿科) 等。EMA的替代终点清单包括授权途径( 常规审批或附条件批准) 、有效成分、适应证、替代终点、关键临床试验、验证等级等信息。我国可以借鉴美国FDA和欧洲EMA 的经验,建立自己的替代终点清单及其COU,列入清单内的替代终点则可以直接作为上市审批的依据,节约研发成本,提高上市审批效率。
3.1.4 建立替代终点认定等级 我国的《技术指导原则》里虽然明确提到了替代终点分为能够合理预测临床获益的指标和很可能预测临床获益的指标,但是尚未建立具体的分类标准和证据级别标准,因此应该借鉴美国FDA 和欧洲EMA的经验,基于替代终点验证的证据级别和风险对替代终点进行分类,经过验证的低风险的替代终点直接运用于标准审批,未完全通过验证风险较高的替代终点运用于附条件批准。并且建立替代终点级别转换制度,可能预测临床获益的替代终点完成验证可以转为能够合理预测临床获益的替代终点,并应用于常规审批。同时,经过风险评价提高了风险等级,或者需要进一步验证的替代终点则进行降级,只能用于附条件批准,或不能作为上市审批依据。这样有利于审批资源的分配,节约审批成本。
3.2 我国基于可能有效的替代终点的新药上市审批程序发展现状及启示
《技术指导原则》里明确提出很可能预测临床获益的替代终点可用于附条件批准。自2018年以来,我国已经通过附条件批准程序批准了多个药品上市,包括阿兹夫定片、帕米帕利胶囊、注射用维迪西妥单抗、普拉替尼胶囊、甲磺酸伏美替尼片、布罗索尤单抗注射液、新型冠状病毒灭活疫苗( Vero 细胞) 、重组新型冠状病毒疫苗( 5 型腺病毒载体) 等。我国已经建立起了针对附条件批准的以《药品管理法》、《药品注册管理办法》、《药品附条件批准上市申请审评审批工作程序( 试行) 》、《药品附条件批准上市技术指导原则( 试行) 》为依托的较完备的法规体系。在《技术指导原则》里对附条件批准的适用范围、沟通交流、上市后要求做出了明确规定。关于我国基于替代终点的附条件批准上市审批路径提出以下建议。
3.2.1 完善技术指导原则 自从加入国际人用药品注册技术协调会 ( The International Council for Harmonisation of Technical Requirements for Pharma- ceuticals for Human Use,ICH) 后,我国的药品注册管理逐渐向国际接轨,最明显的调整就是技术指导原则的架构和完善。目前出台的《技术指导原则》还较为宽泛,主要集中于附条件批准程序上的技术指导,应该针对具体的研发方向和范围制定更为具体的技术指南,为药品的研发过程提供科学的参考和可遵循的路径。加快替代终点药学通用技术指南的起草与修订,为附条件批准上市的药学研究提供充足的技术保障,更好地服务药学研究与技术审评工作[21]。
3.2.2 公开基于替代终点的附条件批准的药品临床试验数据 在美国 FDA和欧洲EMA的网站上可以查询到附条件批准上市药品的详细数据。我国虽然已经建立起了临床试验登记和临床试验结果公开制度,但是关于我国通过附条件批准上市药品的详细数据目前仍未实现公开,无法通过公开数据判别是否是基于替代终点、基于什么替代终点及其作用机制等。建议对基于替代终点的附条件批准的药品临床试验数据给予一定的公开,可以为后续的研发提供参考。
3.2.3 健全再审评程序 我国的药品附条件批准上市执行的是“批准-验证-再审评”的程序,通过这种形式加快具有突出临床价值的临床急需药品上市[22]。建议对再审评的评价依据进行明确,同时借鉴美国FDA的经验,对于申请再审评的通过附条件批准程序上市的药品不仅审核附条件批准中的上市后承诺,还要对风险效益进行全面评价,充分满足常规审批的要求给予正式批准。也可以借鉴欧洲EMA的相关经验,根据不同的情况,给予维持、暂 停、变更或撤销的不同审批结论。

来源:Internet


