您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-10-27 11:14
摘要:目的:探讨阿达木单抗生物类似药相似性评价和适应症外推的药学考量。方法:通过调研国内外已获批阿达木单抗生物类似药申报情况,结合研发和审评实践,对阿达木单抗质量属性风险分级及相似性评价方法进行简要介绍,着重讨论药学相似性评价要素的内容和基本考量,并阐述适应症外推的药学考虑。结果和结论:阿达木单抗质量相似性研究是其整体相似性评价的基础,也是适应症外推的前提;针对药学比对研究中所观察到的候选药和参照药之间的质量差异,应考虑产品质量属性之间的关联性,并结合非临床和/或临床证据科学论证质量差异是否具有临床意义。
2015年我国原国家食品药品监督管理总局发布了《生物类似药研发与评价技术指导原则(试行)》,对生物类似药的研发、评价和管理工作具有重要意义。鉴于生物制品具有相对分子质量大、结构复杂、活性对其结构完整依赖强、生产工艺复杂等特点,为了进一步规范和指导生物类似药开发和评价,结合当前研发和审评实践, 2021年2月国家药品监督管理局(National Medical Products Administration,NMPA)进一步发布了《生物类似药相似性评价和适应症外推技术指导原则》[1]。该指导原则药学内容重点介绍了生物类似药药学研究和评价要素、如何进行生物类似药质量相似性评价、适应症外推时的药学考虑等,为生物类似药药学相似性评价提供更为细化和规范的指导建议。
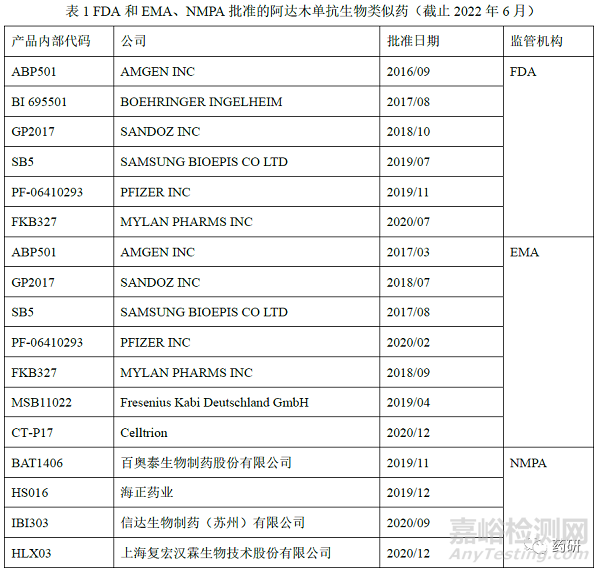
阿达木单抗(adalimumab)的原研药修美乐 (Humira),由Abbvie公司研发,是全球首个获批上市的全人源抗肿瘤坏死因子(Tumor necrosis factor-α, TNF-α)单克隆抗体(IgG1κ),每条重链由451个氨基酸组成(糖基化位点N301),每 条轻链由214个氨基酸组成,分别于2003年、2010年在美国和中国上市。作为生物类似药研发最热门的靶点之一,据统计国内有30余家企业申报阿达木单抗生物类似药的临床研究,自2016年9月美国国家药监局(Food and Drug Administration, FDA)批准Amgen公司申报的Amjevita上市,截止2022年6月FDA、欧洲药品管理局(European Medicines Agency,EMA)、NMPA 和一共批准了近20款阿达木单抗生物类似药(表1)。修美乐上市以来申报获批了多种规格和包装形式,并且进行了处方变更。目前修美乐在全球获批的适应症多达9个,其在不同适应症的作用机制也不尽相同[2],其生物类似药研发和相似性评价具有代表性,因此,本研究以阿达木单抗为案例,深入讨论生物类似药质量相似性评价和适应症外推的药学考量,以期为工业界和监管机构研究和评价提供参考。

一阿达木单抗生物类似药质量相似性研究
1.1 质量属性的评估分级
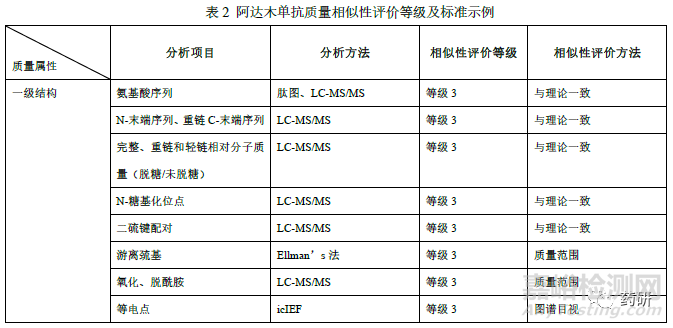
申请人应结合已有研究数据和公开文献等,依据阿达木单抗参照药的质量属性对生物学功能、药动/药效(PK/PD)、安全性、有效性和免疫原性的潜在影响,采用合适的风险评估工具对质量属性进行分级。随着对产品认知的不断深入,进而更新产品关键质量属性与临床风险获益之间的相关性,以及在不同适应症中作用机制不同,也可能对质量属性的分级重新进行调整。本案例中阿达木单抗质量相似性评价指标和方法详见下表(表2),相似性接受标准为,等级1 的质量属性采用等效性检验(产品作用机制或临床表现直接相关的质量属性),等级2 的质量属性采用90%以上候选药的检测结果落在参照药质量范围内(x±3s,临床风险获益具有相对较低风险的属性),等级3 的质量属性为定性或图谱比对(临床风险获益具有较低风险的属性或非定量数据)。对于阿达木单抗生物类似药质量相似性研究不限于本示例中列举的分析项目和分析方法及相似性评价标准。
1.2 质量相似性评价
在质量相似性评价时,应考虑参照药和候选药的选择、方法的确认和验证,以及理化特性、纯度和杂质、生物学活性、免疫学特性及Fc 功能、稳定性相似性比对研究等。
参照药和候选药的选择方面,修美乐在国内先后获批了 40mg:0.8mL 预填充式注射笔、40mg:0.8mL 预填充式注射器、40mg:0.4mL 预填充式注射笔、40mg:0.4mL 预填充式注射器、20mg:0.2mL 等多种规格和包装形式。其中40mg:0.8mL 和40mg:0.4mL、20mg:0.2mL 的规格制剂处方不同,为改善患者的依从性,高浓度规格的修美乐中去除处方中的离子缓冲系统(枸橼酸盐/磷酸盐)、氯化钠等,仅保留甘露醇和聚山梨醇酯80 两种辅料。原则上候选药和参照的规格和包装形式应一致,如不同,应有充足的理由和依据。对于阿达木单抗而言,目前国内已获批的阿达木生物类似药企业均选取40mg:0.8mL 的规格用于相似性研究,但后续正在研发的企业可能面临40mg:0.8mL 规格的不可获得,提醒申请人关注不同规格制剂处方差异可能对产品质量和稳定性的影响。若候选药开发多种规格,相似性评价时应纳入不同规格的制剂;候选药应纳入拟商业化工艺生产的批次,通常包括临床、工艺验证批次等;对于中试规模的批次,如评估工艺变更前后候选药质量具有可比性也可纳入相似性分析。在批次数量上,为充分捕获参照药结构的异质性和批间的变异性以及充分表征和评估候选药的批间质量差异,美国FDA 要求纳入至少10 批次具有一定时间跨度的参照药与6~10 批次的候选药进行质量比对研究[3]。如ABP501 质量相似性评价纳入10:24:18 批(候选药:美国来源原研药:欧盟来源原研药),BI 695501 纳入13:58:86 批,PF-06410293 纳入10:68:78 批。我国指导原则中要求选择的批次具有代表性,而批次数量尚缺乏细化的说明,但国内研发企业纳入的参照药的批次也均达到10 批以上,候选药也至少达到6 批以上。目前我国研发的部分阿达木单抗生物类似药在临床试验期间发生了不同程度工艺优化、规模放大等,其中某生物类似药在临床期间工艺变更前后药学分析显示具有差异,制剂处方也发生了变更,且变更前样品用于PK 相似性研究,申请人开展了包括变更前后样品和参照药3 臂的全面药学比对分析和非临床桥接研究。为提供整体相似性评价一致的物质基础,建议申请人尽早采用拟商业化工艺生产的样品用于相似性研究,特别是关键性临床比对研究。
分析方法的验证和确认方面,用于分析相似性评价的方法应经过验证或确认,其中用于原液和制剂质量标准放行和货架期的检测方法应经过全面的验证。用于检测功能活性或通过图谱比对的表征检测项目[如差示扫描量热法(DSC)、圆二色谱(CD)等])的分析方法,可不进行全面的验证和产品特异性的确认,但应确保其分析方法科学合理、符合预期用途,具有良好的可靠性和重现性。
理化特性分析包括一级结构、高级结构、翻译后修饰(不限于糖基化修饰)研究等。对于采用“头对头”定性比对或图谱比对方式进行评估的质量属性,如氨基酸序列、相对分子质量、二硫键配对、等电点、消光系数、高级结构等的分析,通常仅需采用适当候选药和参照药批次用于比对研究。对于一级结构分析,应通过多种酶切的形式使质谱覆盖率达到100%,且氨基酸序列原则上应与参照药一致。对于高级结构分析,除表中列举的技术手段外,国外企业还采用了较为先进的方法,如氢氘交换、x 射线结晶、核磁共振技术等,有助于证明候选药和参照药的指纹级相似。若候选药和参照药高级结构的表现略有差异(如由于制剂处方的不同,热稳定性指标Tm 值有差异等),申请人应提供相关研究数据论证差异不是产品本身分子间的差异。阿达木单抗的翻译后修饰一般为氧化、脱酰胺/天冬氨酸异构化、糖化、N/C 末端变异体等。氧化位点通常为Met34、 Met83、Met256 等,其中Met256 在FcRn 结合区域,考虑到可能会降低FcRn 亲和力和热稳定性,若氧化比例高于参照药,应结合亲和力检测和稳定性研究(强制降解)进行评价。脱酰胺修饰通常发生在重链Asn27、Asn30、Asn319、Asn388 等位点,应结合修饰位点位置(是否在CDR 区)等评估可能对产品质量的影响。另外还有候选药可能有三硫化物、硫醚、半胱氨酸化修饰等,也应结合修饰的含量及类型评估对产品质量的影响。同时应关注样品处理过程可能对翻译后修饰类型和含量的影响以及稳定性研究期间翻译后修饰的变化。
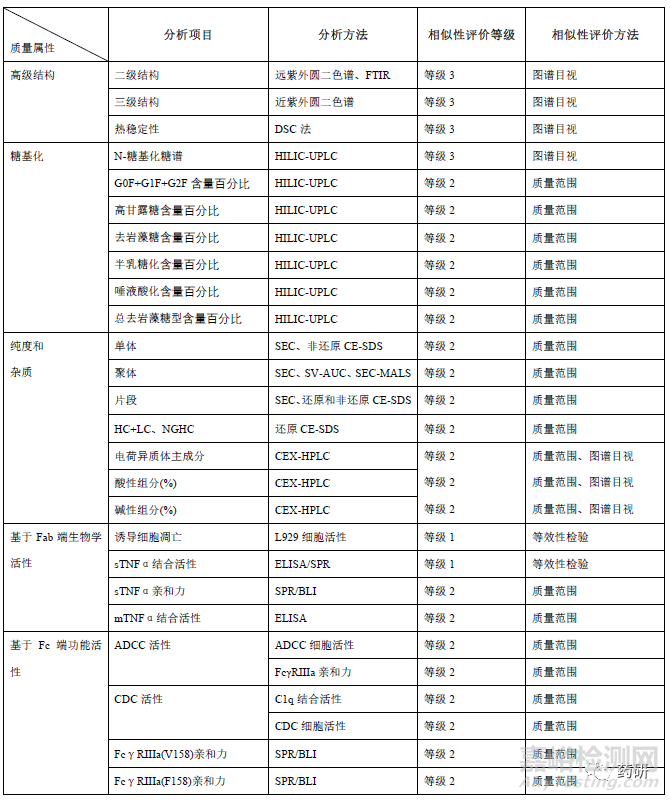
糖基化修饰方面,目前已获批的阿达木单抗生物类似药,均采用 CHO 细胞作为宿主细胞,和参照药一致。糖基化修饰位点均确认为N301,与参照药的糖型谱图均具有相似的分布,但在糖型含量或糖基化占有率上略有差异(表3)。在进行相似性评价时,基于当前对糖型和Fc 端功能活性、PK、免疫原性的关系的认知,一般可将不同糖型按照百分比含量统计为总去岩藻糖(去岩藻糖+总高甘露糖)、去岩藻糖、末端半乳糖、总高甘露糖、总唾液酸等进行分类比较。总去岩藻糖包括G0-N、G0、G1、G2 和高甘露糖型,可增加FcγRIIIa 亲和力以及ADCC 活性;末端半乳糖通过增加抗体和C1q 的结合力增加CDC 效应;高唾液酸可能增加抗体半衰期、减弱单抗的ADCC;高甘露糖型可缩短单抗的半衰期、增强单抗的ADCC 功能。因此,糖基化修饰作为关键质量属性之一,原则上候选药应与参照药具有相似性。若糖型含量存在差异,可结合候选药和参照药的FcγRIIIa、C1q 的亲和力、酶切去除唾液酸前后生物学活性差异、ADCC、CDC活性、临床PK 表现等综合分析。若候选药和参照药的ADCC 活性和CDC 活性、以及临床PK 等均相似,或糖型含量虽有差异,但均较低(如唾液酸糖型含量有差异,但均小于1%),即使糖型分布及含量存在差异也不能排除候选药和参照药的相似性。同时也会关注是否早期开发批次糖型分布含量差异较大,工艺优化后的临床批次和工艺验证批次与参照药更相似。
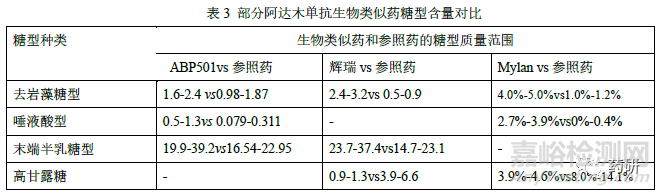
纯度和杂质方面,申请人应尽可能采用先进、正交、灵敏的方法对候选药和参照药的纯度和产品相关杂质进行定量及表征。在对大小异构体进行相似性研究时,通常采用SEC-HPLC、CE-SDS 等检测方法对所有批次进行分析。首先应进行谱图的叠加比对,若有新峰产生,应说明其成分并评估对产品质量的影响。如安进公司ABP501 在SEC 图谱中聚体峰和单体峰之间有新峰,但含量<0.1%,结合其他正交的方法研究评估认为影响可忽略不计。再对单体、重链+轻链、聚体、片段等关键质量属性进行统计学数据分析,若聚体或片段含量有差异,应结合含量多少和差异大小以及生物学活性检测结果等综合评估。另外还应关注此类质量属性随时间推移的变化情况,必要时应分别对时间调整后(有效期末)和未调整(放行时)的结果进行分析。电荷异构体可能影响产品的有效性、安全性和免疫原性,也是候选药和参照药最易产生差异的质量属性之一。应尽可能收集候选药和参照药的各组分馏分并进行表征鉴定,开展不同组分的生物学活性比较;通常阿达木单抗酸性变异体主要为含脱酰胺、氧化、唾液酸化修饰的变异体以及片段等,碱性变异体主要为重链含有1 或2 个赖氨酸的变异体、449 位脯氨酸酰胺化等。评价时可结合CpB 酶切前后的图谱、各组分TNF-α结合和中和活性、ADCC活性等检测结果综合评估电荷异构体的含量分布差异对候选药和参照药的相似性影响。由于候选药和参照药参照药宿主细胞和生产工艺的不同,工艺相关杂质(细胞培养成分、残留HCP、DNA、蛋白A 等)可能和参照药参照药不同,但应评估杂质谱和杂质水平的异同对产品安全性的潜在影响,并制定合理的质量控制策略。
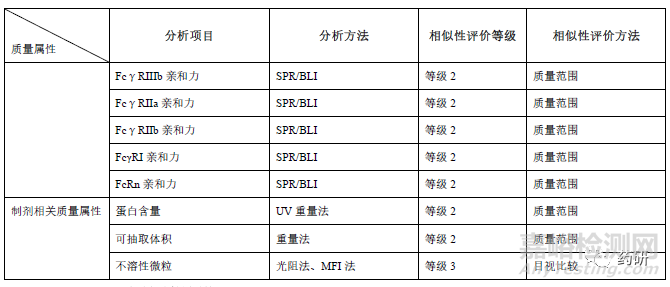
生物学活性和功能研究应采用反映其作用机制的检测方法,并尽可能采用多种不同的生物学活性分析方法对产品进行全面的活性分析和相似性评估。对具有多重生物活性的,其关键活性应当分别进行分析和评估。阻断可溶性TNFα和TNFR 结合被认为是阿达木单抗主要作用机制,通常采用等效性检测评价候选药和参照药的细胞凋亡抑制活性和可溶性TNFα(Soluble TNFα,sTNFα)结合活性(可采用酶联免疫吸附实验/表面等离子体共振/荧光能量共振转移等方法)。另外还评价膜型TNFα结合、反向信号、ADCC(NK 细胞/PBMC)、CDC 等功能活性、以及FcRn 受体、Fcγ受体亲和力(FcγRⅠ、FcγRⅡa、FcγRⅡb/c、FcγRⅢa V158、FcγRⅢa V158 F158)、 FcγRⅢb)、混合淋巴细胞反应(mixedlymphocyte reaction,MLR)、抑制VCAM-1 黏附分子表达、LT-α结合等。生物学活性对糖型含量、电荷异构体等质量属性差异的解释论证至关重要,应尽可能选用经充分优化、变异性小且能反应作用机制的测定方法。若活性测定采用不同批次的参比品,应关注不同批次参比品的生物学活性的桥接标定。
稳定性比对研究是质量相似性研究和评价的重要内容,热稳定性和强制降解对比研究应纳入相似性评价,用于评价候选药和参照药潜在的结构差异和降解途径(杂质)的差异。强制降解研究通常进行包括氧化、强酸、强碱、光照、振荡等条件下的降解谱的比对。由于强制降解研究可能掩盖某些降解机制,应该开展加速(如25℃ 6 个月)稳定性对比研究。若候选药的制剂处方和参照药不一致,可能导致产品降解速率可能不同,应证明产品稳定性差异是源于处方差异而不是分子本身的差异(如当置换为和参照药一样的制剂处方时,结果显示产品降解速率一致)。应关注如果稳定性研究不是“头对头”进行时,方法变异性可能对降解速率差异的影响。另外稳定性研究选择样品的在效期内时间点也可能对比对结果产生影响。长期稳定性研究支持候选药保存条件和有效期的设定,且不能差于参照药;候选药和参照药的有效期可能不一致,如修美乐的有效期为24 个月,而ABP501、SB5 等获批的有效期为30 个月。
一般属性方面,如渗透压、pH、外观、澄清度、颜色、聚山梨酯80 等质量属性的相似性研究,参照药可以纳入适当批次,另外由于制剂处方的不同,上述质量属性可根据制剂处方和批次放行结果,拟定合理的质量标准。制剂相关属性中,蛋白浓度需将所有批次纳入相似性分析中,可抽取体积也尽可能纳入更多的批次,阿达木生物类似药如采用了西林瓶的包装形式,应关注灌装体积和给药剂量的准确性。不溶性微粒分析一般比较亚可见微粒的多少,检测≥2、5、10、25μm 的微粒,还应区分球形和非球形的颗粒;常用的分析技术包括不溶性微粒检测仪、微流成像颗粒分析系统,亚微米颗粒使用动态光散射和场流分离(field-flowfractionation)技术。
其他方面,为规避专利侵权,生物类似药的制剂处方和参照药可能不一致。制剂处方辅料应符合药典标准,如加入新的辅料,应关注对候选药质量和稳定性、临床表现的影响。另外候选药可能采用不同的包装形式,如预填充式注射器、自动注射器等,如仅二级包装不同,且不影响产品质量,也可仅纳入一种规格相似性评估。
二适应症外推的药学考量
质量相似性是整体相似性的基础,也是适应症外推前提,适应症外推应对完整的相似性证据链进行整体评估,药学、非临床及临床的相似性证据之间应相互验证、相互支持。适应症外推时应考虑疾病因素的复杂性,并结合认知的深入进行科学考量。通常使用敏感模型开展临床比对研究的,其结果可用于支持适应症外推。对于作用机制不完全相同的适应症,外推时需额外的研究数据支持。
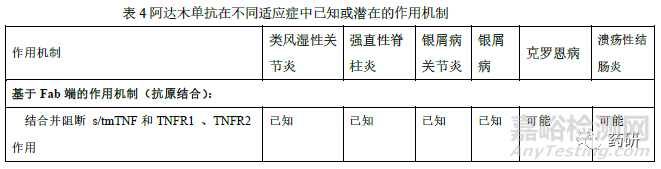
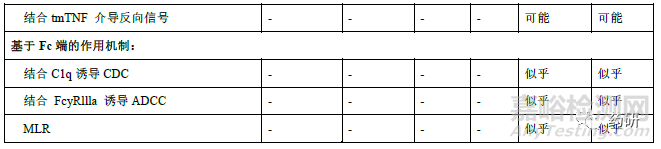
修美乐在国内已获批葡萄膜炎、克罗恩病、类风湿性关节炎、强直性脊柱炎、银屑病和多关节型幼年特发性关节炎等多个适应症。根据阿达木单抗说明书、已发表的研究以及其他TNF 抑制剂在相关疾病中研究的认识,阿达木单抗在不同适应症中已知或潜在的作用机制不尽相同,详见表4。其中类风湿性关节炎、银屑病、强直性脊柱炎、幼年特发性关节炎、化脓性汗腺炎具有共同的TNFα依赖性疾病病理分子机制。sTNFα诱发炎症性肠病(克罗恩病、溃疡性结肠炎)中的炎症反应,但对可能影响炎症性肠病有效性和安全性的作用机制尚未完全清楚。因此,外推至炎症性肠病时,除证明与sTNFα结合和中和TNFα诱导信号传导通路方面的相似性外,还应比较候选药与参照药的ADCC 和CDC、膜型TNFα结合及阻断MLR 中增殖的能力、反向信号研究等,也可考虑建立针对体外炎症性肠病的细胞模型(如抑制HCT-116 细胞凋亡和IL-8 释放)。必要时可调整上述相关生物学活性和功能检测项目的相似性评价的权重和评分。
三讨论
近年来,我国生物类似药研发水平逐渐提高,越来越多的候选药进行中美双报或中美欧多国申报,在开展相似性研究时,可能选择非国内来源的参照药用于非临床或/和临床研究,应开展多臂的药学甚至PK 相似性研究,支持不同国家来源的参照药研究数据的桥接。在国内进行上市申请时,应以中国批准上市的原研
药品作为参照药建立与候选药的质量相似性。生物类似药候选药质量标准应参考ICH Q6B 和《中国药典》等相关指导原则制定合理的检测项目和可接受范围/限度。如果候选药和参照药采用相似的分析方法和分析原理,并且相似性评价认为高度相似的,标准限度建议尽可能与参照药保持一致。对于与临床作用机制直接相关的体外生物学活性项目,尽管采用的检测用细胞系可能和参照药不一致,设定标准的范围应和参照药一致;对于大小变异体的检测项目,一般应至少控制制剂质量标准主峰限度不低于参照药,原液的主峰限度可结合原液的贮存条件和时间、降解情况等合理拟定;对于糖型项目,应结合不同糖型异质体对生物学活性、PK、免疫原性等的潜在影响制定合理的控制水平,由于候选药和参照药在糖型分布和含量方面的差异,可能考虑在过程控制或放行质量标准中额外对关键糖型设定限度/范围或增加ADCC、CDC活性等检测项目。电荷变异体项目一般考虑酸性变异体、碱性变异体和主峰,通常碱性变异体为含有1 或2 个末端赖氨酸的变异体,可结合不同组分鉴定和表征的结果设定总赖氨酸变异体和不同酸性变异体的指标。
TNF 抑制剂在不同疾病中作用机制复杂,尽管尚不确定ADCC、CDC 活性是否与炎症性肠炎的临床有效性直接相关,但在无Fc 功能的的抗体类药物(培塞利珠单抗)中观察到有效性降低[4];同时在相似性分析时,也应关注活性检测体外模型细胞不能代表病理条件下体内细胞的局限性,如Celltrion 公司在比较英夫利西生物类似药CT-P13 和参照药的功能活性时,只有在体外构建的膜型TNFα高表达的细胞中才能检测到ADCC 活性。因此,在进行适应症外推时,还应结合候选药与参照药的非临床研究、临床研究比对结果综合评价质量属性的差异对产品的免疫原性、PK、安全性和有效性的影响。
生物类似药候选药上市后按照产品生命周期管理,当发生药学变更时(如引入新的包装形式、规格等),应参考ICH Q5E 和《已上市生物制品药学变更研究技术指导原则(试行)》相关指导原则开展研究。但考虑到生物类似药可能的质量漂移,上市后增加参照药新获批的适应症时,应对其上市后积累的生产批次进行质量分析,重点对与适应症外推相关的关键产品质量属性进行回顾总结,以评估产品质量对适应症外推的支持程度。
我国生物类似药的研发起步较晚,近年来国家不断制定和颁布多项鼓励研发的相关政策,促进生物类似药市场的发展,《生物类似药相似性评价和适应症外推技术指导原则》的发布,可进一步规范和指导国内生物类似药的药学相似性研究和评价。本文以阿达木单抗为例,介绍了质量相似性研究和评价相关内容,供工业界、研发者及监管机构交流讨论。

来源:Internet


