您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-10-29 05:14
我国2020年版《药品注册管理办法》中提出了突破性治疗药物程序、附条件批准程序、优先审评审批程序和特别审批程序四种药品加快上市程序,针对药品附条件批准上市程序,国家药品监督管理局又发布了相关配套文件《药品附条件批准上市申请审评审批工作程序(试行)》和《药品附条件批准上市技术指导原则(试行)》,为企业申请附条件批准上市提供了明确的条件、程序及要求等依据,药物临床试验期间,治疗严重危及生命且尚无有效治疗手段的疾病以及公共卫生方面急需的药品,药物临床试验已有数据显示疗效并能预测其临床价值的,以及应对重大突发公共卫生事件急需的疫苗或者国家卫生健康委员会认定急需的其他疫苗,经评估获益大于风险的,可以在上市许可申时提出附条件批准。附条件批准上市的目的是缩短药物临床试验的研发时间,使其尽早应用于无法继续等待的危重疾病或公共卫生方面急需的患者。
本文通过分析国内的药品附条件批准上市申请审评审批工作程序、要求以及实施过程中的问题,借鉴美国的加速审批与欧盟附条件上市许可在药品附条件批准上市申请审评审批方面的程序和经验,以期能够完善我国药品附件批准上市申请审评审批工作程序和技术要求,进一步加快临床急需的创新药上市,体现“以患者为中心”的药品审评体系导向。
中国、美国、欧盟附条件批准政策的对比详见表1。



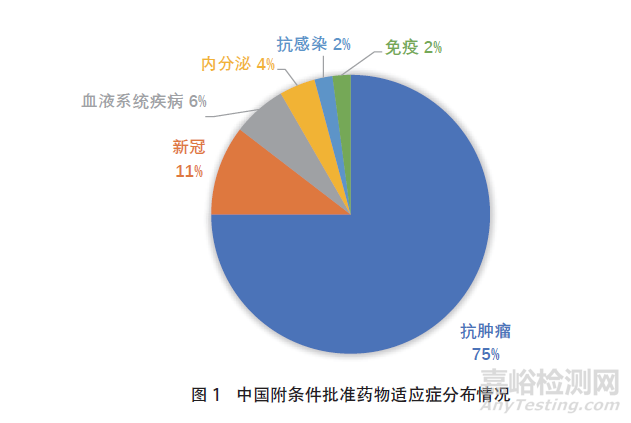
截至2021年12月31日,中国药品附条件批准情况见图1。
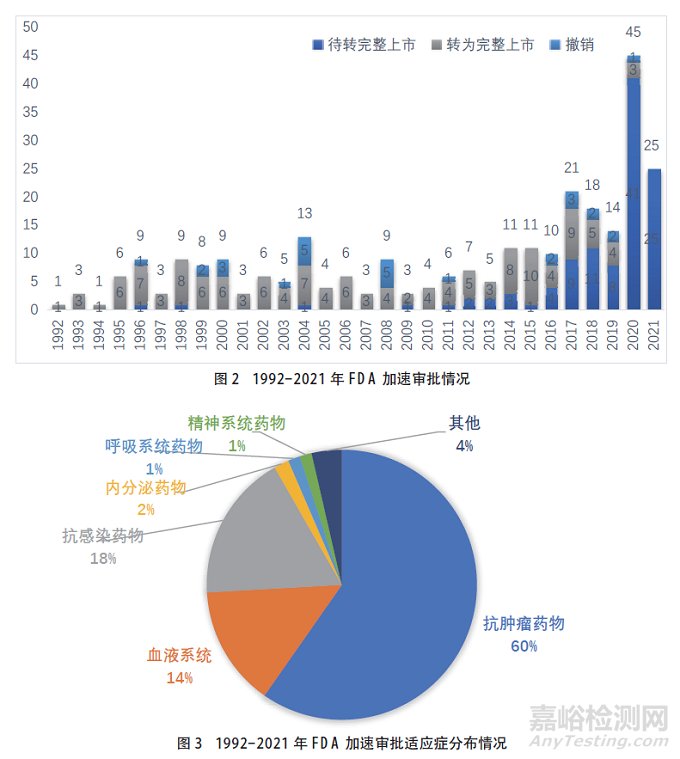
1992~2021年美国FDA药品附条件批准情况见图2。加速审批产品情况见图3。
欧盟药品附条件批准情况见图4。
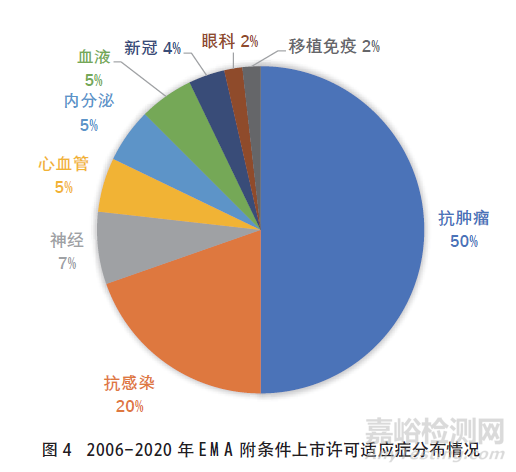
在药品的适应症分布方面,中国附条件批准上市药物涉及的适应症领域不及美国和欧盟广泛,其中抗肿瘤药物占比最高。在中国、美国和欧盟附条件批准上市的药物中,虽然均是抗肿瘤药物占比最大,但中国抗肿瘤药物占附条件批准上市药物的比例最大,约为75%,欧盟占比最小,约为50%,美国FDA占比约为60%。
在附条件批准上市转为常规批准(完全上市)方面,从上述数据可以看出中国附条件批准转为常规批准的转化率是10.41%,美国是50%,欧盟是38.98%,当然这与中国实施附条件批准时间较短有关,后续药品监管机构应持续关注附条件批准品种上市后的研究工作以及补充申请申报情况。
在附条件批准上市的注销方面,自FDA正式设立附条件批准程序,已有28个附条件批准被撤销资格,FDA对于附条件批准上市品种注销有具体情形以及具体流程。欧盟基于延续注册申请下对特定义务开展的定期评估和基于申请人未履行义务下的被动评估,也涵盖了附条件上市许可产品的调出情形。而中国对附条件批准上市的注销有规定,但尚未明确具体情形以及程序,仅是要求持有人在规定时限内以补充申请形式提交上市后研究资料。跨学科审核支持;三是可以滚动提交资料。
附条件批准的目的是在风险可控的前提下,缩短药物的研发和注册时间,使可能有临床获益的药物尽早应用于患者。其带给申请人机遇的同时也带来挑战,需要申请人对疾病背景和治疗领域有深入的理解,并且对药物的研发具有前瞻性。同时政府部门需要规范审评尺度,完善审评路径,加强上市后监管,引导申请人全面认识附条件批准的要求,平衡风险与获益,更好地促进药物创新,加快药物的研发和上市速度,为患者提供更多有价值的药品。
参考美欧附条件上市的法规政策,对国内附条件批准政策有以下几点思考。
《药品审评质量管理规范:突破性治疗认定药品和生物制品的管理》提到考虑撤销BTD认定的几种情形包括:一是临床数据不再显示相比于现有治疗有显著提高;二是相同适应症的其他药物获得批准上市并且认定突破治疗药物临床获益没有很大提高;三是认定突破治疗药物不遵循研发计划。
一是关于附条件批准药品治疗疾病的类型。我国2020年发布的《药品附条件批准上市技术指导原则(试行)》中已对附条件批准的适用范围作出了详细说明,即用于严重危及生命且尚无有效治疗手段的疾病、公共卫生方面急需的药品和应对重大突发公共卫生事件的疫苗。严重危及生命的疾病是指若不尽早进行治疗会在数月或更短时间内导致患者死亡的疾病或疾病的某个阶段,例如晚期恶性肿瘤等。但指导原则并未对适应症进行限定,在中国、美国和欧盟附条件批准上市的药物中,虽然均是以抗肿瘤药物为主,但中国抗肿瘤药物在附条件批准上市药物的占比最大。同时可以注意到非严重危及生命的药品,如用于治疗系统性红斑狼疮的注射用泰它西普也已通过附条件批准上市。
附条件批准是为鼓励以临床价值为导向的药物创新,加快具有突出临床价值的临床急需药品上市。建议附条件批准对疾病严重性和药品急需性进行综合考量,监管机构需要帮助申请人对附条件批准的适用范围进行更深入的理解,适当开发抗肿瘤药物以外的其他适应症药物。
二是关于替代终点、中间临床终点的选择。在药品的有效性评价方面,对于符合附条件批准情形的药品,可基于替代终点、中间临床终点或者早期临床试验数据而附条件批准上市。替代终点的选择必须科学合理,应能显示其与临床获益的关联性和生物学合理性。近年来,对于很多疾病,特别是肿瘤已经建立了如无进展生存期、客观缓解率等替代指标。为了帮助申请人选择潜在的替代终点,促进产品开发,FDA会根据最新的科学研究和审评考量,每6个月更新发布推荐或认可的替代终点。目前国内的指导原则中已对替代终点、中间临床终点的选择方向进行说明,但根据科学进展,针对具体疾病的推荐终点可能更有助于申请人在临床研究设计时参考。
三是完善附条件批准的沟通交流。附条件批准的沟通交流涉及早期临床研究、上市申请前、审评期间三个阶段,为申请人与监管机构之前提供了良好的信息交流桥梁。在时限上,除同时纳入突破性治疗的药物外,目前与附条件批准相关的沟通交流会议均为II类会议,一般在申请后60个工作日内召开。与其他在研发关键阶段申请沟通交流的药物时限一致。为加快药物上市,可以考虑缩短附条件批准的沟通交流的时限。此外,由于附条件批准药物有大量研究工作是在上市后开展,可以考虑在上市后沟通交流的程序并在时限上提供政策支持,以帮助药品尽早常规批准上市。
四是完善审评路径,做好与《疫苗管理法》紧急授权的衔接。《疫苗管理法》第二十条明确,应对重大突发公共卫生事件急需的疫苗或者国务院卫生健康主管部门认定急需的其他疫苗,经评估获益大于风险的,国务院药品监督管理部门可以附条件批准疫苗注册申请。出现特别重大突发公共卫生事件或者其他严重威胁公众健康的紧急事件,国务院卫生健康主管部门根据传染病预防、控制需要提出紧急使用疫苗的建议,经国务院药品监督管理部门组织论证同意后可以在一定范围和期限内紧急使用。在《疫苗管理法》中,疫苗的加速上市存在附条件批准和紧急使用授权两种途径。在施行时存在疫苗产品获批紧急使用后申请附条件批准的情形,以智飞生物的重组新冠病毒蛋白疫苗为例,其于2021年3月10日在国内获批紧急使用,于2022年3月1日在国内获批附条件批准,但目前紧急使用授权与附条件批准之间的衔接程序还有待明确。
五是加强上市后监管。虽然中国、美国、欧盟附条件上市政策中都会要求申请人在药物获批上市以后,继续完成上市后临床试验,并在规定时间内提交这些临床试验的相关资料。然而有报告指出,在美国有些申请人会有意或无意地拖延提交上市后临床试验资料,使得这些通过附条件批准的产品既不满足完全批准要求,又未被撤销,却能够继续留在市场上。因此,国内监管部门如何确保在药品上市后,企业在规定时间内按要求完成临床试验仍然是一项挑战。我国目前出台的法规政策中,对于附条件批准的注销情形和程序尚不清晰,仅笼统描述“逾期未按照要求完成研究或者不能证明其获益大于风险,国家药品监督管理局应当依法处理,直至注销药品注册证书”是远远不够的。建议明确附条件批准注销具体情形和程序。

来源:《中国药事》


