您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-12-02 04:04
● 什么是可比性研究(CP)?
● 为什么要设定这个指导原则?
● CP总体要求是什么?
● 变更CP的内容,FDA有什么建议?
● 提交了CP并得到批准之后,在执行过程中出现了变化,怎么办?
● 执行CP以及执行之后还要提交什么?
药品在其生命周期中往往为了追求更好的质量控制或者更高的经济效益,可能会对CMC部分进行各种各样的变更。这些变更最终是否能够成功得到监管机构的认同和批准,可比性研究(comparability protocols)在其中发挥了至关重要的作用。为了更好的指导企业正确处理其商业化过程中的变更,并更有效的同监管机构沟通,FDA在2022年10月发布了相应的指导原则。本文希望能通过康日百奥CMC团队过往多个商业化项目的变更以及与监管机构沟通的经验,解读FDA最新发布的这份指导原则,从而帮助业界同仁能更顺利的完成商业化阶段的变更和可比性研究。
2022年10月FDA发布了指导原则,Comparability Protocols for Post-approval Changes to the Chemistry, Manufacturing, and Controls Information in an NDA, ANDA, or BLA. (可比性研究指导原则,以下简称“CP”),这一指导原则适用于已获得或即将获得NDA或BLA批准的药物,企业在如何处理商业化过程中发生的变更、如何就这些变更和监管机构进行效率化的沟通、如何不影响到产品的商业化进程,给出了一定的指导意见。
药物在即将商业化的过程中,企业对药物CMC的设计方向从最大程度保证临床试验的成功,而逐渐转为如何最大化商业利益。除去销售团队的智慧和运营外,CMC团队最大的挑战来自于如何在保持产品质量不变的前提下最大程度降低COGs(Cost of Goods)。往往在NDA/BLA之前,企业已经在考虑是否自建产能或者委托CMO生产,是否要将生产规模提升到2000L甚至更高,是否可以使蛋白的表达水平或者纯化回收率进一步改善,是否可以使用同等质量的国产物料进行替代等等。这些都会带来批准后的变更(post-approval change)。CP对这些变更的申报提供了一种解决方式,康日百奥团队将通过此文章向大家介绍并解读这一指导原则的具体内容。
1 什么是可比性研究?
绝大部分企业,即使是常年征战美国市场的企业,对于comparability protocols不会太熟悉,反而大家会对“post approval change”也就是“批准后变更”比较熟悉。
在指导原则中,FDA提出了CP的概念:
A CP is a comprehensive, prospectively written plan for assessing the effect of a proposed Post-approval CMC change(s) on the identity, strength, quality, purity, and potency of a drug product, including a biological product, as these factors may relate to the safety or effectiveness of the product.
从上面这段话不难看出,CP是即将实施的CMC变更的评估和计划。在GMP系统中,CP相当于变更前提交给QA或者变更委员会(CC)的材料。
接下来指导原则中提到CP可以包括在NDA/BLA中,也可以在批准后提出。企业可以在BLA中提交CP,将后续计划的变更前置,为自己争取了时间。
2 为什么要设定这个指导原则?
随着中国企业逐渐走进欧美市场,NDA、BLA和ANDA慢慢地被FDA所批准,批准后的变更无法回避,就会涉及到向官方提交变更,提交变更的方式有PAS,CBE-30,CBE-0和annual report。那CP和这些缩写是什么关系呢?
先简单介绍变更申请的方式。从变更的风险程度看,重大变更风险程度最大,一般涉及到场地的变化,或者化学药杂质谱的变化,通过采用PAS申请方式,涉及的变更需要FDA仔细审评批准后企业方可实施;CBE-30和CBE-0申请方式适用于中等变更,风险程度中等的变更,给FDA递交材料后30天内或者递交当天,如果官方没有反馈,变更即可执行;annual report申请使用于微小变更,一般每年固定时间给FDA提交资料,该类变更的风险程度较低。变更类型及申请方式参见下表。
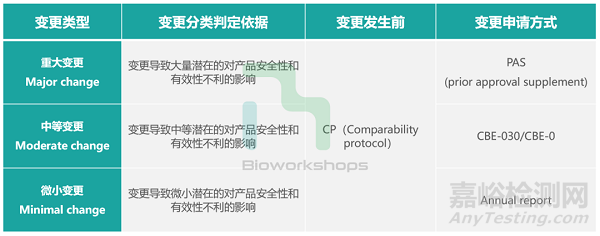
企业要不要做这个变更,除客观因素以外,官方对于变更的分级也非常地重要。谁都不希望出现内部判定成CBE-0或者CBE-30的变更被FDA升级成PAS。但最终结论往往要到变更提交后才会尘埃落定。
CP的优势是将变更通知前置。CP适用于所有的变更,在变更发生之前递交变更计划,风险评估。企业按照官方批准后的变更计划、风险评估和申请方式进行变更。重大变更、中等变更和微小变更的CP可以直接在原申请的3.2.R中递交,下文中有具体描述。变更往往不是单一存在的,多个变更同时存在或变更导致的变更等情况相对很复杂,企业在认知上存在差异性,对复杂变更把握的水平稂莠不齐,提前与FDA确定变更计划、风险评估和申请方式,可极大的减少企业试错的经济成本和时间成本。
除了上面提及的目的之外,指导原则还提到了申请变更方式的降级。如果变更执行之后产品质量没有受到不良影响(这个结论的最终确定并不简单的,下文会提及),那原本可能是PAS的变更会被降级成CBE-30,CBE-30会被降级成CBE-0,等等。这种降级申请/通知方式给产品后续的商业化生产和销售带来利好消息,意味着质量等同但经济效益更高的药品会更快地进入市场。
3 CP总体要求是什么?
CP的总体要求高。指导原则中提到:A CP describes the specific tests and studies to be performed and the acceptance criteria to be achieved to demonstrate the lack of adverse effect of one or more proposed CMC changes on product quality.
在变更前要开展评估试验,并提出评判变更前后产品质量等同的接受标准。并且如果想降低变更的级别,企业需要提出基于科学和技术的说明用于支持降低变更对产品质量的风险。关于产品的说明从何而来?指导原则提供了六个方向,包括经验、产品特有的属性、产品研发的数据、工艺验证和专门的支持性试验数据:
● 历史经验(Prior Knowledge);
● 原液特性和生产工艺的研究开发过程;
● 成品特性、制剂处方和制剂生产工艺研究开发;
● 工艺验证,包括工艺设计、首次工艺确认和持续的工艺确认;
● 质量风险控制活动;
● 在缩小规模上的变更执行,用于模拟商业化规模上的变更执行。
当向FDA提交CP时,指导原则推荐CP应该包括一个描述性的标题、版本号、日期等用于追踪信息;CP隶属于CTD 模块 3的3.2.R章节(即对应中国CTD格式中的3.2.R.5 可比性方案)。执行变更后,如果变更均按照CP中的计划执行,并且达到了CP中预定的可接受标准,可以按照CP中的描述降级申请。这里指导原则提到,对于一些复杂的变更(比如:改变制剂浓度、改变制剂处方等),需要非临床或者临床等效性数据的支持,这样的变更不适用CP。
4 变更CP的内容,FDA有什么建议?
FDA想看见什么?作为一个长期和监管机构打交道,并参与过许多不同品种工艺转移的注册,本人有机会看过不少国际大公司的注册文件,从中获得了一个很深的体会:注册文件不在于囊括工艺参数的多与少和开展试验的多与少,最终是否能通过审评或者拿到clinical hold缺陷的多少,除了要满足指导原则的要求和行业内的一般认知这个大前提之外,更重要的在于是否提供了审评员所重点关注的信息:IND期间首要重点永远是安全性,当产品逐步走向NDA/BLA时审评重点会逐渐向产品的有效性倾斜。这就不难理解,FDA审评员这几年会格外关注IND阶段项目的原辅料来源、工艺中的内毒素和微生物负荷控制、in-use stability等。
言归正传,康日百奥团队接下来详细解读一下CP所需包括的内容。官方推荐提交表格化的信息、描述性的语句和/或图表对以下方面进行总结和概括。(建议GMP体系的变更提交同样参照以下要求。)
1 变更描述(变更是什么?)
详细说明变更以及变更的背景。
2 支持性的信息和数据(为什么FDA同意批准?)
提交的信息和数据应该能够说明申请人对变更的理解,包括变更可能对产品和工艺产生的风险以及控制风险的策略。一般会提交以下信息:
● 以往的变更经验;
● 针对计划变更的风险评估总结;
● 基于产品研发过程的数据,从科学性和技术性角度出发,预测变更对于产品质量的影响;可以在缩小模型/小规模上进行相应的研究以丰富支持评估的数据;
● 其他试验,用于增加申请人对于变更的理解和认知;
● 风险控制策略的说明。
3 可比性方案(变更前后产品质量怎样判定等同?)
明确受到影响的产品,可以是原材料、中间过程物料(比如化学药物酰化反应结束后的反应物,或者生物药纯化工艺阴离子层析后的收集液)、中间品、原液、制剂、内包材等,头对头地对比变更前后产品质量(不仅包括受到影响的物料,还包括工艺后续的DS和DP);一般使用商业规模的DS或/和DP进行比较,需要说明产品的批号、规模、生产场地和生产工艺。
基于风险评估,确定需要开展的对比试验。可以是日常的放行检测、中控测试和稳定性考察。但是,一般不认为这些日常测试能够充分控制变更的风险。因此要增加结构表征的相关测试,用于评估变更的风险。另外,稳定性研究,比如强制降解或者影响因素,也可以考虑是否也需要纳入可比性研究。
需要描述分析方法,这些分析方法能够用于评估变更并保证产品质量。分析方法包括样品测试方法、取样方法和统计学方法,科学地对变更前后的产品质量进行对比。
变更前后的产品首先要符合既定的质量标准,同时也要达到上述为评估变更而设计试验的目标;也要符合统计学判定质量等同的标准。
如果是质量标准发生变更,CP方案应该说明变更后的质量标准能够提供与原标准等同的或更好的质量控制。
4 变更结束后的申请级别(以哪种形式提交给FDA?)
FDA推荐降级汇报,比如CBE-30,CBE-0或者年报形式,比如原本是PAS的变更可降级为CBE-30或CBE-0。但是对于厂房变更,新的厂房需要FDA开展审计,这种情况可能无法降级。
5 其他信息
在其他信息中可以提交:
● 说明CP是一次性的变更,还是会重复发生的变更;
● 提供申明:The site will not distribute product manufactured with the change(s) until the site’s quality control unit has confirmed that the acceptance criteria specified in the protocol have been achieved and has approved the implementation of the change;
● 提供变更执行的时间等。
5 如何处理批准后的变化?
FDA提供了一连串的21CFR用于说明变更的“变更”是有法可依的,感兴趣的朋友可以点击此处查看原文。值得一提的是,指导原则举出一些变更的“变更”的例子:
1 如果发生了以下变更的“变更”,采用CBE-30
● 对批准的CP结构表征试验进行替代或者更改;
● 对批准的CP中可接受标准进行变更,认为变更后的可接受标准可以满足相同的质量控制要求;
● 同时执行几个已批准的CP;
2 如果发生了以下变更的“变更”,采用CBE-0
● 对批准的CP结构表征试验进行替代或者更改,明确可以提供更好的控制策略;
● 增加测试或者增加可接受标准。
3 如果发生了以下变更的“变更”,采用年报形式
● 按照官方要求/药典要求改变结构表征测试、分析方法或者可接受标准;
● 缩紧可接受标准;
● 按照批准的CP对另一个CP中的信息进行改变;
● 仅文字编辑。
6 执行CP以及执行之后还要提交什么?
当执行变更后,申请人应该回顾下最初的风险评估,确保风险评估的有效性。如果评估的风险被改变了,这可能会影响到申请的级别。如果出现这种情况,及时和FDA沟通。
当完成变更后,如果数据符合CP方案中既定的质量标准,按照CP方案中的是申请级别向FDA提交文件,即降级申请;如果数据不符合接受标准,或者在变更中发生了一些意料之外的事件,FDA建议申请人及时和FDA沟通,来确定如何申报这些变更。
指导原则到这里基本告一段落,将变更可比性的前期提交和后期提交的整个流程介绍清楚。整体看来,变更的风险评估和开展的试验结果是重点,用于降低官方对变更后产品质量可比性的担忧;在执行过程中,需要按照方案去执行,尽量不要去随意改变变更的内容,最终向FDA递交变更执行的申请,为变更画上完美的句号。
不得不说,指导原则侧重的是较强的叙述逻辑以及广泛的适用性和覆盖面,这一原则也同样适用于注册文件的撰写。注册人员从FDA发布的指导原则书写习惯中择善而从。
最后借着FDA指导原则的原文,着重强调下:Regardless of the type of change, the methods used in, and the facilities and controls used for, the manufacture processing, packing, or holding of a product, including packaging and labelling operations, testing, and quality control of products, must comply with current good manufacturing practice (CGMP). CGMP是评估变更和执行变更的基石,尤其在场地变更中,cGMP体系将是现场检查的关键。
产品NDA/BLA的批准,不是研发/注册的中止,而是新旅途的开始。

来源:同写意


