您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-12-05 00:57
FDA有权随时对生产FDA监管产品的制药公司进行检查,并将其意见记录在FDA 483表格上,通常缩写为“483”。实际上,不仅是制药公司,临床试验机构、医疗设备公司、CDMO,都可能随时受到FDA 的随机和例行检查。 FDA 监管食品、药品、疫苗、医疗器械、化妆品和烟草产品。一家制药公司所能承担的最大风险之一,就是其产品质量行为遭受FDA 的质疑。当产品不能在美国销售,尤其是拳头产品(blockbuster drug)遭受影响时,这家制药公司可能瞬间陷入困顿。
当 FDA 进行检查时,他们可以提前提醒公司,也可能突击出现。检查结束后,FDA 会准备一份差异清单,并将发送FDA 483表格,然后可能根据检查观察结果发送警告信(所以FDA 483 表格不是警告信,两者之间既有关联,又是有区别的)。
483 表格对于受 FDA 监管的公司来说非常重要。一个 483表可以准确地告诉公司哪些方面需要改进或纠正,以从偏离正轨的道路上重新回归,并保持合规性。淡化解读483 的内容,或者更致命的是,不认真不完整地回应483的观察结果,可能会产生代价昂贵的后果。有点类似足球比赛中对于裁判的口头警告不以为然,依旧我行我素地毫无收敛,那就离红黄牌不远了。
调查人员对可能违反法规的观察结果被记录进483。根据FDA调查人员所做观察的数量和严重程度,483可能会对被检查的制药公司或医疗设备制造商造成严重后果。收入损失是明面的影响,而改进合规流程所花费的时间,以及声誉受损则是 483 表格和警告信潜在的灾难性影响。
什么是 FDA 表格 483 观察?
FDA表格483观察,也称为“inspectional observation”或“483 表”(也有“缺陷报告”的意译称呼),是FDA检查人员在检查过程中发现的任何潜在违规行为。483 表可能与公司的设施、设备、流程、控制、产品、员工实践或记录有关。
当调查人员观察到违反《食品药品和化妆品法案》及相关法案的任何情况时,在检查结束时都向接受检查的公司管理层发出FDA 483 表格。FDA检察人员都受过专门的培训,以确保483表中的每一项观察记录都明确、具体而且重要。当观察到任何食品、药物、设备或化妆品的质量问题,或者正在准备、包装或保存可能影响质量从而导致损害患者(用户)健康的产品时,FDA检查人员就会在483表格上记录下该项观察。
FDA 483 表格的用途是什么?
483表 的目的是通知公司管理层所观察到的负面情况。检查结束时,调查人员将提交483 表,并与公司高级管理层讨论。在收到 483 表后,公司有 15 天的时间做出回应。FDA鼓励公司以书面形式回应483 表,列举出他们相对应的纠正措施计划,然后迅速实施该这些纠正措施计划。
483 表格并不是对违规行为的最终解释,但它们代表着公司亟待关注和解决的问题。483 表的签发可能会使公司损失数百万美元。 483 表可以触发培训、重新设计、工艺实施和其他措施。发出 483表 后,FDA 可能会认为受监管机构存在严重违规行为,此时警告信就会发出。
FDA 483 表格对机构执法有何影响?接下来会发生什么?
FDA 表格 483 并不构成对是否违反 FD&C 法案(联邦食品、药品和化妆品法案, Federal Food, Drug, and Cosmetic Act)或其任何相关法规的最终决定。 FDA 483 表格、一份称为机构检查报告的书面报告、现场收集的所有证据或文件,以及公司做出的任何回应,都将被监管机构列入最终的考虑范畴之中。FDA在考虑了所有这些信息之后,才会确定采取何种进一步行动来保护公众健康。FDA的执法流程序列如图1所示:
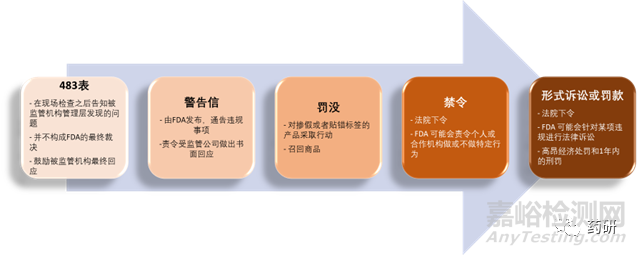
图1. FDA执法流程图(图片来源:eleapsoftware.com)
483 观察的最常见原因
未完全遵循程序
制药行业必须建立标准操作程序 (SOP) 并及时更新它们,以提供合规、保证质量和安全的证据。 SOP 记录必须位于中心位置并可供调查人员随时获取。当员工没有得到准确或最新的书面指示时,或者如果他们没有明确遵守书面指示,错误和质量问题肯定会发生。
对差异或故障的调查不力
需要彻底生产过程中的各类偏差 (deviations),并记录这些调查的结果。如果公司无法确定造成变差或者OOS的根本原因(root causes),而且没有进行充分记录,则 FDA 认为其内部调查不完整。未能调查质量事件可能会给制药公司带来各种后果。
缺少书面程序
FDA 的483观察中经常会记录制药公司缺少书面程序的问题。公司没有遵循有关监管行业的经验法则,也就是记录所做的,按照记录规定行为。许多公司都在管理和控制书面程序的问题上出现纰漏,因为这可能非常耗时并且需要付出额外的努力。如果各种偏差没有得到及时、充分和有效的记录,就不可能查明根本原因并采取适当的纠正和预防措施(CAPA)。
数据完整性问题
数据完整性已成为生命科学行业的普遍问题。 FDA 和 WHO 最近发布了他们的数据完整性指南。这是一个常见问题,但很容易消除。FDA 在过去一年的检查中发现数据完整性问题超过 125 次,例如数据在计算机系统中不安全的问题。公司必须保护电子或物理记录。
清洁、消毒和维护
调查过程中经常观察到制造设施中的清洁问题。正确清洁和消毒设备对于获得优质产品至关重要。如果没有制定和遵守设备的调整、清洁和其他维护计划,FDA 可以提出意见。
环境监测
许多公司没有认真对待环境监测,尤其是制造设施。但 FDA 对此非常重视,并希望对无菌设施进行环境监测。必须在设施的所有分类区域制定和实施适当的环境监测计划。环境监测必须按照预定的时间间隔进行,并且这个监测本身也应该受到监测。
如何解决这些问题?
· 定期进行预防性模拟审核,以了解组织需要关注的重点。
· 遵循 CAPA(纠正和预防措施)计划。此过程遵循模拟或真实检查,以解决任何不合格情况的根本原因。
· 保持最新和准确的 SOP(标准操作程序)。 SOP旨在实现效率、质量输出和性能一致性,同时减少沟通不畅和不遵守行业法规的情况。
· 相应地保护和备份数字和物理记录。
· 对制造设施和质量控制实验室中发生的每一次不合规格、不合趋势、事件偏差或故障的事件进行调查。每个失败的批次或测试都要彻底调查根本原因,即使该批次未放行分发时也应如此。
· 保持所有设施和设备的适当清洁、卫生和维护。
· 执行适合行业的环境监测。
收到 FDA 483 表观察后该怎么做
如果收到了 FDA 483 表观察,需要高度重视小心处理。在检察员离开机构之前,必须与检查员一起审查该文件。应该透彻理解观察结果,以及需要的改正措施。
在检查员离开之前,应务必:
· 了解所记录的观察结果并确保其准确性
· 了解 FDA 发出观察背后的更广泛的信息
· 识别并讨论观察中的任何错误
· 提问
· 展示对适用法规的认识
· 必要时咨询法律顾问
公司应在 15 天内发送书面回复,否则 FDA 将无需在决定后续行动时考虑公司的回复。提交书面回复后,FDA 将审查 483 表观察结果、企业检查报告和回复。FDA将使用此信息来确定要采取的进一步行动。
警告信
在签发 483 表格且检查员完成机构检查报告后,监管机构可能会发出警告信 (这并不取决于公司是否做出书面回复,当然不做书面回复可能会收到警告信)。当发现产品质量存在严重违规行为时,上级主管部门在审查完意见后可以发出警告信。警告信明确表示,公司必须采取行动纠正问题,并为该组织的行为方式提供了指示和时间表。
FDA 483 表格和警告信有什么区别?
483 表格是通知公司可能存在违规行为的通知,而警告信是该通知的严重升级。公司必须在收到 483 表和警告信后 15 天内应以书面形式回复 FDA。
483表格不是对违规行为的最终裁定,它们是需要紧急解决的问题,而警告信是当质量和合规性被认为不可接受时发出的官方缺陷信。并非所有 483 项观察结果都会产生警告信,但公司必须做出回应并且必须愿意遵守 FDA 规定。

来源:药研


