前言
近年来大家关注的主流灭菌方案有美国注射剂协会发布的《湿热灭菌程序的验证:灭菌程序的设计、开发、确认及日常监控》、 欧盟推出的《制剂、原料药、辅料和内包材灭菌指南》、日本厚生省的《通过最终灭菌方法生产无菌药物的准则》及中国的《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》。
针对如此繁多的灭菌指导原则,本文就和大家聊聊如何在符合申报条件下选择更具有科学性和优势的灭菌方案?以及灭菌到底在灭些什么?终端灭菌注射剂最终走向何方?
一、 无菌药品
1.1 无菌药品的概念
《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》针对无菌药品给出了准确的概述,即是指法定药品标准中列有无菌检查项目的制剂和原料药,一般包括注射剂、无菌原料药及滴眼剂等。
由于目前无菌检验手段的局限性,不存在绝对意义的无菌,现在大家所理解的无菌只是概率意义上的“无菌”,即采用科学合理的SAL(无菌保证水平)保证最终产品的PUSN(无菌保证值)达到≤10E-6。
1.2 两个重要概念
先和大家聊聊比较容易混淆两个概念,即SAL和PUSN:
SAL(英文全称:Sterility Assurance Level):无菌保证水平,通过科学合理的灭菌/除菌工艺、良好的无菌保证体系及严格执行药品生产质量管理规范( GMP )使药品中活微生物存在的概率低至某个可接受的水平,SAL的主体是灭菌/除菌工艺。
PNSU(英文全称:Probabilityofa Nonsterile Unit):无菌保证值,是指通过科学合理的灭菌/除菌工艺,制剂产品的无菌保证值必须≤10-6,PNSU的主体是产品。
1.3 本文涉及的范围
面对如此纷繁复杂的无菌技术,本文仅涉及采用终端灭菌工艺的最终灭菌产品,以点破面,来了解各国无菌策略的异同。
二、 主流国家的灭菌策略
2.1 美国注射剂协会
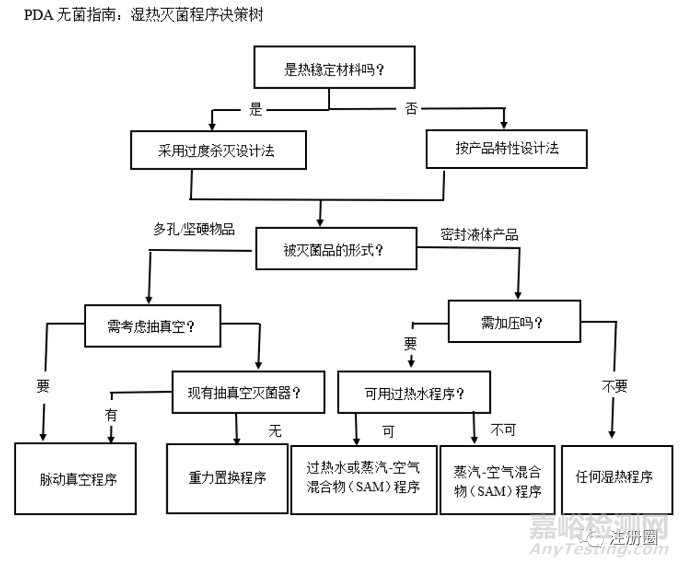
美国注射剂协会(PDA)发布的《湿热灭菌程序的验证:灭菌程序的设计、开发、确认及日常监控》,PDA的理念是根据灭菌物品热稳定性科学合理的选择相适应的灭菌程序。
PDA的无菌指南分为过度杀灭和根据产品特性设计灭菌这两大灭菌策略,过度杀灭程序强调的是被灭菌产品获得F0PHY( 物理杀灭时间) 和F0BIO (生物杀灭时间)至少为12 min ,根据产品特性设计灭菌程序强调的是针对不同种类产品,科学合理的选择不同的灭菌方式;虽然也有过度杀灭程序,但是不纠结F0和8min的关系。
2.2 欧盟
欧盟推出的《制剂、原料药、辅料和内包材灭菌指南》,以水溶性产品的灭菌决策树终端灭菌工艺为例,欧盟的理念是尽可能地提高生产工艺的无菌保证水平,用以保证产品的无菌保证值。
欧盟的无菌指南强调的是灭菌条件首选121℃,15min,其次就是选择F0≥8min,SAL≤10-6,提出了产品各组份可以根据产品特性分别科学合理的选择不同的灭菌方式,尽可能地提高生产工艺的无菌保证水平。
2.3 日本
日本厚生省的《通过最终灭菌方法生产无菌药物的准则》,日本的理念是更加注重对终端热处理的应用。
日本的无菌指南强调的是F0值<8min,即热处理工艺的运用,利用生物指示剂和微生物负载结合法,科学合理的选择更高的无菌保证水平,此无菌策略需要进行严格的灭菌验证及日常微生物(物料及环境)负荷检测,这个和没有灭菌选择决策树是日本的无菌指南的两大特点,所以大家现阶段在仿制日本注射剂需要特别注意其灭菌方式,尽量避开不符合国内无菌指南的产品。
2.4 中国
国内的《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》,基本上借鉴并简化了欧盟的《制剂、原料药、辅料和内包材灭菌指南》。
国内的无菌指导原则强调的是过度杀灭法(F0≥12min)和残存概率法(min 8≤F0),这两种灭菌工艺不合适只能选择无菌生产工艺,并没参照PDA及日本指南,在F0<8min的热处理上使无菌保证水平达到≤10-6,这样简单粗暴的灭菌工艺选择有好有坏。
好处:
(1)降低制药企业灭菌工艺选择的难度;
坏处:
(1)相当一部分的注射剂无法仿制,尤其参比制剂是日本厂家的;
(2)一部分的注射剂一致性评价无法进行,F0值<8min的;
(3)会降低注射剂的无菌保证水平,终端热处理变成无菌生产工艺,带来临床安全问题;
(4)昂贵的生产成本及更低的无菌保证水平,不利于国内产品出口;
三、 如何在符合申报条件下选择更具有科学性和优势的灭菌方案?
3.1 仿制药灭菌方案
查询参比制剂处方工艺或经参比制剂重复灭菌推测,选择与参比制剂相同且不低于国内无菌指导原则的灭菌方案,如日本相当多的注射剂采用F0<8min的终端热处理方案,国内现阶段对于F0<8min的终端热处理方案不认可,若采用残存概率法(min 8≤F0),产品的理化指标容易超参比制剂且不符合质量标准,采用无菌生产工艺又难以达到参比制剂的无菌保证值,所以在调研仿制项目时需考量参比制剂的无菌保证水平即灭菌/除菌工艺是否符合国内的无菌指导原则。
3.2 创新药灭菌方案
创新药的灭菌方案应依据各组分的化学结构特点、处方工艺研究及理化指标稳定性,以患者的临床收益为核心,现阶段按国内无菌指导原则(注射剂灭菌工艺选择的决策树)科学合理的选择更具有科学性和优势的灭菌方案。
四、 灭菌到底在灭些什么?
灭菌通俗的讲就是通过科学合理的灭菌/除菌工艺、良好的无菌保证体系及严格执行药品生产质量管理规范( GMP )使药品中活微生物存在的概率低至某个可接受的水平,即这个可接受的水平必须≤10-6。
灭菌的核心是患者的临床收益,科学合理的灭菌是根据产品特性设计灭菌程序,是针对不同种类产品选择不同的灭菌方式,在产品的无菌保证值和质量之间寻找一个平衡点,即在满足产品无菌保证值的前提下使用一切可以使用的灭菌/除菌工艺来提高产品质量,尽最大可能提高患者的临床收益。
灭菌到底在灭些什么?灭菌为患者灭掉了安全风险,留下了药品疗效。
五、 终端灭菌注射剂最终走向何方?
我推测现阶段国家发布的《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》是基于当前的认知的过渡性指导原则。
我们身处技术大爆炸时代,随着国内科技水平的提高,对灭菌和无菌工艺研究更加的深入,以前的灭菌/无菌策略经验在以后不再全盘适用,不再拘泥于简单的灭菌决策树,更多基于患者临床收益为核心的灭菌/无菌策略必将涌现。
终端灭菌注射剂最终走向何方?
当前:采用国内无菌指导原则的灭菌方案;
以后:会综合采用基于产品特性的灭菌方案,即对不同种类产品,科学合理的选择不同的灭菌方式,模糊F0和8min的关系,终端热处理工艺也将会被考虑使用;
未来:随着对灭菌和无菌工艺研究的深入(尽最大可能提高患者临床收益),全套设备系统性能的提升(更高的无菌保证水平,可以全程即时的微生物负荷水平监测),自动化迈入新的阶段(消除人为的干扰),会更加科学合理的设计灭菌/无菌工艺(针对性的设计产品独有的灭菌/无菌工艺)。
六、 总结
以患者的临床收益为核心是我们制药工作者的出发点和落脚点,现行的灭菌/无菌工艺带来的临床安全问题会随着时间的推移逐渐显现,这可能也是国家基于当前的认知做出的无奈之举。
相信随着国内科技水平的提高,我国的无菌指南会综合学习并改进发达国家的无菌策略,更加科学合理的选择产品的无菌工艺,保障患者的临床收益。
参考文献
[1] 《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》
[2] 《中国药典》(2020 年版)
[3] 国家药品监督管理局.关于发布除菌过滤技术及应用指南等3 个指南的通告 (2018 年第 85 号)
[4] 谢纪珍, 冯巧巧, 刘军田,等. 化学药品注射剂灭菌工艺选择及工艺验证常见问题探讨[J]. 药学研究, 2018, 37(6):3.
[5]邹萍. 化学药品注射剂灭菌工艺选择和工艺验证常见问题分析[J]. 神州, 2019(18):1.
[6]杨森森, 李永丰, 魏俊卿,等. 化学药品注射剂灭菌工艺选择及工艺验证中注意事项[J]. 益寿宝典, 2020(028):000.
[7] 《通过最终灭菌方法生产无菌药物的准则》
[8]PDA Sterilizing Filtration of Liquids Report No. 26 (2008Revision) of PDA.
[9] PDA Process Simulation Testing for Aseptically Filled Productsechnical Report No. 22 (2011 Revision) of PDA.
[10] PDA technical Report No. 44 Quality risk management foraseptic processes(2008).
[11] EU.GMP Annex 1 : Manufacture of Sterile Products(2020).
[12] EMA. Guideline on the sterilisation of the medicinal product,active substance, excipient and primary container(2019).