目的:为制药企业了解新版《药品生产质量管理规范》(GMP)的新要求,做好洁净室的改造工作提供参考。方法:在对比新、旧版GMP洁净要求的基础上,分析洁净室改造的考虑要点,并提出建议。结果与结论:新版GMP在洁净区级别、悬浮粒子测量、送风口风速、同一洁净级别的相邻房间的压力差等方面与旧版有所不同。压力差、送风量与回风量、气流组织形式、送风口出风风速、换气次数是洁净室改造的考虑要点。为此,洁净室改造要分改变风口数量和不改变风口数量2种情况进行设计。
新版《药品生产质量管理规范》(GMP)终于在大家的翘首期盼下于2011年3月1日开始实施,其主要沿用了欧盟GMP的框架,并且采用了很多欧盟GMP中的规定,这将对我国很多制药企业造成很大的影响。新版GMP中对于洁净室洁净级别的重新划分和定义,将使得制药企业不得不思考,如何进行洁净室的改造才能符合其要求,本文将就此问题进行探讨。
1、新版GMP洁净要求概述及与旧版的区别
1.1 新版GMP对洁净区的划分及要求
新版GMP将无菌药品生产洁净区分4个级别[1],具体如下:
A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。单向流系统在其工作区域必须均匀送风,风速为0.36~0.54 m·s-1(指导值)。应当有数据证明单向流的状态并经过验证。在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低的操作步骤的洁净区。
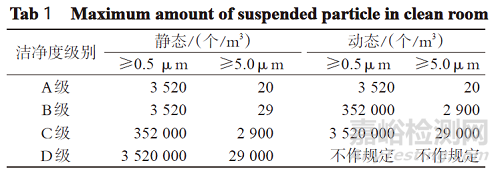
以上各级洁净区的悬浮粒子最大允许数详见表1。
表1 各级洁净区悬浮粒子最大允许数
1.2 新旧版GMP对洁净区要求的主要区别[2]
①新版GMP将洁净区分为A、B、C、D 4 个级别,而1998 年版GMP是三十万级、十万级、万级、百级4 个级别,并且新版的4 个级别要求更加严格,能够更好地保证药品的质量。
②新版GMP对悬浮粒子的测量首次引入动态测量的标准。1998 年版GMP只是规定了一种状态下的悬浮粒子要求,即企业通常执行的静态标准;而新版GMP要求不仅在静态下测量的悬浮粒子达到相应要求,更是要求在设备运转额定动态下也能达到相应的要求。
③新版GMP的A级洁净区与1998 年版的百级洁净区一样,虽然都是属于层流区域,但是高效过滤器送风口的风速要求却完全不一样。新版GMP规定的风速为0.36~0.54 m·s-1,但是在密闭的隔离操作器或手套箱内可以使用较低的风速;而2003 年版《药品生产验证指南》中推荐的风速为≥0.25 m·s-1。
④新版GMP规定同一洁净级别的相邻房间的压力差不得小于10 Pa,洁净室与洁净走廊之间的压力差不得小于10 Pa。相比1998年版GMP,同一洁净级别的相邻房间的压力差增加了1倍,对于空调净化系统和房间设计提出了更为严格的要求。
2、洁净室改造的考虑要点
正是由于新版GMP对洁净室更新更高的环境要求,很多制药企业在规定的时间内必须进行洁净室的改造或者在新建洁净室时考虑更多的因素来满足新版GMP的要求。
2.1 压力差、送风量与回风量
新版GMP将不同洁净级别之间的相邻房间的压力差提高到了10 Pa,这将大大提高对空调净化系统的运行能力的要求。高压力差的规定就要增大新风量,增加空调净化系统的负荷,缩短过滤器的寿命。
压力差[3]是由送入新风量的大小来维持,也可以说压力差是由送风量减去回风量来维持的,其建立的基本原理是送风量大于回风量、排风量、漏风量之和。其中,送风量、回风量以及排风量都是由空调净化系统决定,而漏风量则是由房间的密封性决定的,所以良好的建筑设计是保证压力差的基础。
如上所说维持洁净室压力差的是风量,所以在实际洁净室的设计和建造中不可能直接设计和控制压力差,而是设计在一定的压力差下的具体风量,因此应该考虑如何进行风量的计算。洁净室压力差风量的计算方法一般有2 种[3]:换气次数法和缝隙法,又因缝隙法既考虑洁净室围护结构气密性又考虑维持室内压力差控制值所需风量,因此比换气次数法更合理、准确。
根据洁净室维持的压力差,为了维持该压力差所需的风量可参考文献[3]公式进行计算。
2.2 气流组织形式
气流组织形式是保证洁净室洁净级别的重要因素和关键的影响因素,一般洁净室的气流组织形式选择是根据洁净室的洁净度而定的[4]。高洁净级别,如A级的气流组织形式一般都是单向层流,而稍低级别的气流组织形式几乎都采用顶送侧回,陈旧的顶送顶回的方式已经不再采用了。
气流组织形式在理论上是由GMP的要求决定的,但是在实际的设计和形成过程中主要是通过送风口和回风口的位置决定的。当然,房间中的设备和工作人员将会影响到气流组织形式,所以在设计设备安装位置时也要考虑到不影响气流流形,并且尽量减少工人数量以及活动行为和范围。
2.3 送风口出风风速
新版GMP对于A级洁净区的风速进行了规定,但是对于其他级别洁净区的风速却没有明确的要求,而出风口风速的大小影响着气流的流通速度,进而影响单位时间的进风量,最终将影响洁净室的送风量、压力差等重要的参数。
由于不同洁净室的洁净级别和体积不同,所以对于风量和风速要求都不相同,但是由于几乎所有的送风口的风速都是由总的空调系统的风机决定的,所以要通过调节风速来调节风量等参数是不可能实现的,必须通过增加和减少送风口和回风口的数量才能实现。
2.4 换气次数
换气次数[5]是指单位时间内通入房间内的空气量相当于房间体积的倍数,单位为次/h。换气次数是决定乱流洁净室洁净度水平的最重要参数。随着换气次数的增加,洁净度级别也相应增加。
对于各个级别的洁净区换气次数,要根据GMP规定的悬浮粒子浓度来计算,计算公式可参考有关文献[6]。对于计算出来的换气次数,如果其值小于10 次/h,那么换气次数的取值是10 次/h;如果其值大于10 次/h,那么换气次数就根据计算出来的实际值作为要求来设计洁净室和安排送、回风口的数量以及位置[6]。
3、改造建议
由于最终的洁净度是所有影响因素共同作用的结果,所以在新版GMP的要求下进行洁净室的改造要综合考虑上述提到的各种影响因素。笔者认为要增加换气次数,回风口的回风量要增大或者回风的频率也要提高,所以以下的讨论都在此假设的前提条件下进行。
在洁净室改造中应该考虑的是在保持现有气流组织形式的前提下,提高进风量、减少回风量,而要达到这样的目的,最容易想到的是增加进风口、减少回风口。但是回风口减少有可能会提高洁净室受到微生物污染的概率,并且风口数量的变化势必会改变风口之间的相对位置,相对位置的改变又将改变气流组织形式,进而有可能造成洁净室的某些区域成为死角,洁净风无法达到该区域,从而成为污染源。如此循环往复必将陷入死胡同,无法理清头绪,所以笔者认为要借鉴自然科学研究的方法——单变量分析法,变化某个因素来考虑其对因变量的影响,从而得出相关的规律。鉴于此,笔者分如下2种情形进行讨论:
(1)如果改变风口的数量如增加或者减少送风口或者回风口,就将引起上述的不利因素,那么就要考虑增加或者减少的风口对气流组织形式不产生影响或者影响较小,如新增的送风口能够与原有的风口共同向一个回风口送风,即在以回风口为中心的两侧设置送风口,以便在保留原有气流组织形式的基础上增加送风量。
(2)如果在不改变风口数量的前提下要增加风量以及换气次数,就要考虑更改电机,提高电机的功率,在相同的时间里,使送入洁净室的气体体积增加。但是更换电机有最高上限的限制,只能达到设定的最大值就不能再改变了。在此方案下,由于风口的数量没有变化,那么换气次数就没有改变或者改变不大,所以洁净室的洁净度无法达到现有4 种级别以及10 Pa压力差的要求。
在改造或者新建洁净室时,一定要先确定该洁净室内要安装的设备形状、尺寸、安装位置以及人员操作所在位置等,因为这些因素都将影响气流组织形式以及洁净风最终到达的区域范围。有些设备的形状或者尺寸会对洁净风形成“阻碍墙”,从而阻挡洁净风充满整个洁净区域。而操作人员的频繁走动会产生更多的污染微粒,并且操作人员的运动和操作所在的位置也会对气流组织形式产生影响。
由于新版GMP对于洁净室的洁净度有了动态的概念,那就要求我们在考虑洁净区硬件提升的同时,加强对软件特别是对人员的要求,在洁净室管理的软件系统上要有所升级。因为如果没有系统的软件进行管理,再好的硬件也无法发挥其固有的性能。
综上所述,洁净室的任何一个改变都可能牵扯到其他若干个参数的改变,洁净室的改造就像一个连锁反应,牵一发而动全身。所以在对洁净室进行改造设计时一定要综合考虑各种影响因素,通过精确的计算和周密的考虑,找到一个洁净室改造的最佳平衡点。完成改造之后,再进行完整系统的人员培训,从而达到新版GMP的洁净室要求。
参考文献
[ 1 ] 卫生部.药品生产质量管理规范(2010 年修订)[EB/OL].
[ 2 ] 梁毅.新版GMP教程[M].第1 版.北京:中国医药科技出版社,2011:75-80、84-85.
[ 3 ] 秦峰.制药企业HVAC系统洁净室的压差控制[J].机电信息,2010,14:20.
[ 4 ] 王颖.谈谈医药厂房洁净室设计[J].科技资讯,2007,9:149.
[ 5 ] 吕洪浩.影响医药工业洁净室洁净度因素的应用分析[J].天津药学,2011,23(1):68.
[ 6 ] 翁念慈.B 级洁净室换气次数的计算与分析[J].机电信息,2010,5:12.