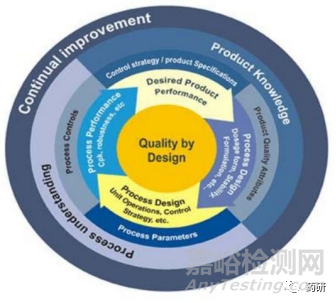
FDA于2004年开始在制药行业倡导质量源于设计(QbD)理念,QbD以预先设定的药品质量为目标,通过充分的科学证据及质量风险管理,并强调对产品、过程的理解以及过程控制,将质量融入到产品的设计、生产、质量提升乃至全生命周期的各个环节之中,从而有效保障药品质量,提高生产效率[1]。
图1 QbD总览图[1]
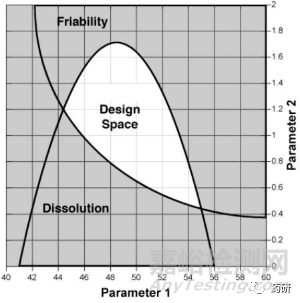
QbD的实施涉及到目标产品概况(TPP)、产品关键质量属性(CQAs)、关键物料属性(CMA)和关键工艺参数(CPP)的分析与确定、设计空间(DSp)的确定、控制策略的形成等环节,而DSp是保证QbD理念实施的一种高效而重要的工具。ICH Q8指导原则将设计空间(DSp)定义为“经过科学论证的物料属性与工艺参数的多维度组合、相互作用范围,在此范围内药品的质量能够得到保证”[2]。药品开发过程中,DSp受到药品监管的审核、批准与监管,在DSp范围内运行,不认为是对关键工艺参数或物料属性的变更,因此,建立药品生产工艺DSp,能够在保证产品质量的基础上,赋予药企最佳的操作灵活性,有利于提高生产效率。
图2 参数重叠形成的设计空间示意图[2]
FDA CDER 的Erik Read等人[3]指出,DSp近年来已经在小分子药物、生物大分子药物生产领域得到逐步应用,其建立涉及风险分析、实验设计(DOE)、数据分析、模型建立、设定值分析、优化等核心环节,每一环节的向下进行都要基于合理科学的数据解读与理解,逐步推进DSp的建立与完善。然而在制药行业缺乏统一的策略与标准,加之DOE执行过程中存在多种变异因素,例如实验测量误差、参数设定范围等,因此导致DSp的建立过程变得复杂而受阻。
Erik Read等人[3]在马里兰州隶属于FDA OBP的 White Oak Facility开展了实验,通过单克隆抗体mAb生产工艺DSp建立过程中实际遇到的4种典型实例情形,阐述如何提高DOE数据解析的准确性与逻辑性,科学引导DSp的建立。mAb生产工艺DSp建立之前,首先确定mAb的CQAs,并预先制定标准限度,接下来通过基于经验、文献资料的风险分析,确定有可能对CQAs存在潜在影响的工艺参数(PP),执行实验设计(DOE)并进行数据分析,根据PP对不同CQAs的影响数据建立拟合模型,并通过拟合模型指标,例如R2、Q2等模型评价指标判断模型的显著性与有效性。根据CQAs是否符合标准限度以及模型的显著性进行情形分类,分为以下4类:
1.CQAs符合标准限度/模型显著
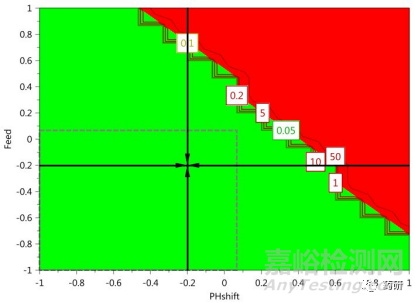
这是DSp建立过程中最理想的情形。这种情形下,在所研究的参数范围内,所生产mAb的特定CQAs均能符合标准限度,而且拟合模型能够反映关键工艺参数与产品关键质量属性之间的联系,具有较强的预测性。这种情况下,甚至不用经过进一步的设定值分析与优化,便可以很容易在此基础上建立DSp。但是需要进一步分析所建立DSp的失败概率,例如,这种情形下,实际建立DSp的失败概率高达50%,原则上,需要最大可能降低DSp的失败概率,确保有效性与可靠性,因此,在此基础上进一步进行了响应曲面(RSM)优化,最终建立的DSp的失败概率降低至10%。由此可见,即使在此理想情形下,也存在进一步优化DSp的必要性。
图3 对CPP进一步优化后DSp的失败概率显著降低[3]
2.CQAs符合标准限度/模型不显著
这种情形下,在所研究的参数范围内,所生产mAb的特定CQAs均能符合标准限度,但拟合模型不能够反映关键工艺参数与产品关键质量属性之间的联系,预测性较差。产生原因往往是由于DOE数据不可靠,例如实验变异因素的干扰。这一点可通过DOE进行过程中并列设计的对照实验结果进行判定,平行实验结果较差的批间一致性,表明了在DOE执行过程中存在未知的变异因素。由于特定CQAs均能符合标准限度,因此可在此基础上进一步进行响应曲面(RSM)优化来完善DSp,提高可靠性。事实表明,经过进一步优化,DSp的失败概率由100% 降至5%。
图4 对CPP进一步优化后DSp的失败概率显著降低[3]
3.CQAs不符合标准限度/模型显著
这种情形下,在所研究的参数范围内,所生产mAb的特定CQAs不符合标准限度,但是拟合模型是显著的,能够反映关键工艺参数与产品关键质量属性之间的联系,具有较强的预测性。显然,出现上述情形的原因是工艺参数范围选取不当,导致mAb的特定CQAs超出了限度。但是拟合模型是有效的,能够对工艺参数对应的CQAs进行有效预测,因此,这种情形下需要对工艺参数的设定值范围进行适度调整,进而建立DSp。经过设定值验证重新建立的DSp的失败概率由100% 降至5%。
图5 经设定值验证优化后DSp的失败概率显著降低[3]
4.CQAs不符合标准限度/模型不显著
这种属于DSp建立过程中最不理想的一种情形。mAb的特定CQAs不符合标准限度,并且拟合模型不显著,主要原因是风险分析不到位,导致潜在关键工艺参数选取不当,以至于不能得到显著的拟合模型。其他原因还包括实验中的其他变异因素、实验误差等。这种情形下,不建议在此基础上建立DSp,而是需要重新谨慎选择实验工艺参数进行实验以建立DSp。
Erik Read等人[3]通过上述 4种DSp建立过程的实例情形,阐述了在DSp建立的过程中如何对实验设计(DOE)的数据进行合理解析,例如CQAs是否符合预定的标准限度、拟合模型是否显著,并在此基础上关注实验过程中的变异因素、潜在工艺参数的选择是否合理、工艺参数范围是否得当,根据上述方面进行决策、下定结论,并分析初步建立的DSp是否需要进一步的优化,例如是否有必要进一步优化以继续降低失败概率,最终成功建立起能够保证产品质量的DSp。
通过这一举措,可以明晰DSp建立过程中的典型模式,并按照这4种模式,合理引导DOE数据的解析,理清DSp的建立策略,降低过程的复杂性,最终成功建立生产工艺设计空间(DSp),促进质量源于设计(QbD)理念在制药领域的实施。