2 0 0 0 年起, 欧盟着手对经分散审批程序(DCP)和互认审批程序(MRP)的药品质量状况进行市场监督抽检。该抽检计划最初由欧盟官方检验实验室主要协作网(GEON)自发组织,经过多年实践,逐步形成了以欧洲药品质量管理局(EDQM)为核心的过程管理体系,以欧盟医药管理局首脑协作组(HMA)为核心的风险管控路径,有效促进了各成员国检验与执法体系的互联共享。截至2021年,欧盟共完成抽检DCP/MRP品种14500余个。作为集中审批药品(CAPs)抽检的重要补充,DCP/MRP药品市场监督抽检旨在对欧盟全境DCP/MRP药品质量进行风险监测,与我国省级药品抽检功能定位相似。因此,研究DCP/MRP药品市场监督抽检策略,对进一步完善我国省级药品抽检管理机制,探索国抽与省抽统筹管理模式,具有一定借鉴意义。
一、法规框架
欧盟议会和理事会颁布的相关指令,是DCP/MRP药品市场监督抽检的主体法规框架。其中,《关于人用药品的欧盟法典》和《兽用药品欧盟法典》,要求各成员国药品监管部门对上市后药品质量进行监督检查;同时,规定各级官方检验实验室(OMCL)建立协调网络,确保检验结果共享互认。在此基础上,欧盟逐步确立了DCP/MRP药品市场监督抽检机制。基于主体法规框架要求,欧盟制定了DCP/MRP药品市场监督抽检的管理程序。一是《DCP/MRP药品市场监督抽检协作规范》,介绍了DCP/MRP药品市场监督抽检工作原则、主要流程,以及组织管理框架。二是《基于风险的市场监督检验网络协作规范》,对上市后药品的质量风险来源进行分类,并提出了基于GEON的抽样与检验风险管理策略。三是《打击药品犯罪协定》与《药品质量安全犯罪与假劣药品处置合作规范》,建立了各成员国协同防控质量风险的管理机制。为保证检验结果的有效互认与准确性,欧盟还要求承检实验室应符合官方实验室质量管理体系规范,并制定了相应的质量管理指南。
二、组织结构与抽检过程管理
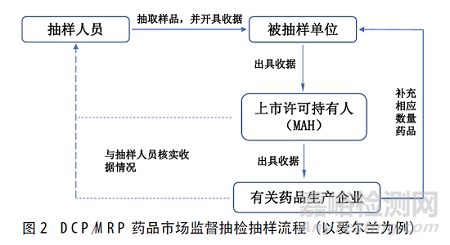
DCP/MRP药品市场监督抽检以EDQM和HMA为核心,基于DCP/MRP药品检验数据库(以下简称数据库)和互联沟通系统(CTS)进行(图1)。其中,EDQM作为DCP/MRP药品市场监督抽检秘书处,主要负责数据库的管理维护、抽样与检验过程管理、检验结果汇总。HMA作为各成员国药品管理部门的综合协作组织,主要负责对发现的质量风险进行综合防控。数据库为抽检计划与数据管理的主要平台,主要提供抽检计划与数据储存、质量风险信息汇总报送,以及在线制图与数据统计功能。在使用权限方面,主要分为两级,各承检的官方检验实验室具有1级权限,可直接对数据进行报送修改;各成员国药品监管人员、药物警戒研究人员、GMP/GDP检查员等具有CTS访问权限的用户具有2级权限,只能对数据进行浏览。此外,EDQM要求各承检OMCLs设置抽检联系人,以“点对点的方式”通过数据库报送抽检计划与检验结果。
三、风险处置与转化应用
对于抽检发现的一般性技术与方法学问题,OMCLs通过检验协作网络或DCP/MRP审评的组织协调机构—人用与兽用药品分散与互认审批协作组(CMDh/CMDv),反馈给参与该品种审评的药品管理机构;对于发现的标准问题,则发送给EDQM下设的标准管理部门。
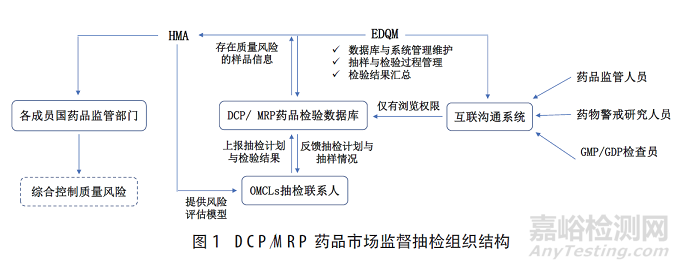
针对检出的假劣药品,各成员国按照《药品质量安全犯罪与假劣药品处置合作规范》,建立由药品管理部门、警察执法部门、司法机关组成的单点联系工作组(SPOCs)。工作组中每个部门都应设置一位决策联系人,定期组织联系会议、协同处置假劣药品。决策联系人应当具有医药相关专业背景和资深管理执法经验。对于涉及多个国家的风险品种,各成员国通过HMA执法人员工作组(WGEO)进行综合协调,并以SPOCs的方式设置国际协调联系人,进行联合执法、制定风险控制措施(图2)。
随着抽检数据与风险信号的积累,基于发现的问题,GEON分别对特定品种进行市场监测研究(MSS)与潜在假劣药品市场监测研究(MSSIP),促进抽检研究结果进一步转化。其中,MSS主要选取在欧盟不同成员国可能出现质量差异的品种进行比较分析,并将提升行业标准的建议反馈至EDQM标准管理部门。总结近5年EDQM年度报告,除2020年受疫情影响外,完成MSS的数量基本在2项以上,提示抽检研究成果转化链条较为稳定,形成了良性反馈与循环。
2 0 1 2 年,基于《打击药品犯罪协定》,欧盟各成员国OMCLs通过GEON组织MSSIP专项研究,旨在充分评估疑似存在假劣药品的供应链风险状况。截至2022年,各成员国OMCLs共完成5项研究,分别涉及合成代谢类固醇、勃起功能障碍治疗药物、减肥药品以及未被WHO统一命名的药物活性成分(“non-INN”APIs)。与MSS不同的是,MSSIP的样品来源范围更广。在由各成员国OMCLs或药品管理部门直接抽取样品的基础上,MSSIP还接受来自医生、患者以及执法机关认为存在质量风险的样品。随机抽样与靶向收样相结合的研究模式,有助于最大范围收集风险信号,更为系统全面地评估供应链安全状况。
四、评价与思考
法规体系:基于管理通则,实现了CAPs年度抽检与DCP/MRP药品市场监督抽检风险管理体系的有机统筹依托《关于人用药品的欧盟法典》与《兽用药品欧盟法典》这一法规基础,欧盟DCP/MRP药品市场监督抽检相关的制度体系主要可分为四个方面:一是过程管理规范,《DCP/MRP药品市场监督抽检协作规范》介绍了DCP/MRP药品市场监督抽检的工作原则与组织框架,并对计划制定、样品抽取、检验结果评估与反馈等方面,提出了总体性的指导要求;二是风险管理通则,EDQM通过制定《基于风险的市场监督检验网络协作规范》,对境内上市后的CAPs与DCP/MRP药品质量风险进行统一归纳、分类,在对GEON提出抽样与检验风险管理策略的基础上,也为HMA组织制定品种遴选风险评估模型,实现CAPs年度抽检与DCP/MRP药品市场监督抽检风险管理体系的有机统一,提供制度基础与管理建议;三是假劣药品处置要求,欧盟于2008年制定《药品质量安全犯罪与假劣药品处置合作规范》,并在2010年签署了《打击药品犯罪协定》,提出了综合防控假劣药品风险的主体要求,为建立横跨多个国家、多个部门的执法协调机制提供了制度基础。因此,可参考欧盟经验,统筹挖掘国家与省级药品抽检数据,对质量风险进行综合分类,制定统一的风险评估指导通则与量化模型,从制度体系上进一步促进国抽与省抽风险管理体系有机融合。
计划管理:建立互评机制,提高抽检的靶向性与协作性、集约监管资源按程序要求,DCP/MRP药品市场监督抽检基于DCP/MRP药品检验数据库,建立了计划在线互评制度。承检OMCLs完成计划制定后,会上传至数据库内公示。根据互评意见,OMCLs可随时对抽检计划进行调整,从而避免重复、过度抽检,充分利用监管资源。此外,通过与CTS建立信息沟通渠道,实现了CAPs抽检与DCP/MRP药品市场监督抽检数据的互联互通,促进数据挖掘、利用的统筹管理。目前,虽然我国已对省级药品抽检结果数据进行统一的管理、分析与利用,但尚未从国家层面建立统一的计划管理平台。建议参考欧盟经验,将管理端口前移,依托国家药品抽检信息系统建立省抽计划统一上传模块与互评机制,协同管理国抽与省抽计划,促进监管资源高效利用。
检验管理:制定统一的检验质量管理体系,可为综合利用抽检结果提供技术支撑欧盟对于检验质量的管理,主要基于两个层面:一个是基础层面,官方实验室质量管理体系规范,对欧盟全境药品检验实验室提出了最基础也最为重要的管理要求;另一个是执行细则,根据GEON的过程管理特点,EDQM制定了较为完备的质量管理指南,从细节上对检验过程中的质量保障、样品登记流转、检验分析、结果研判与上报进行规范指导。统一的质量管理要求,为不同药品质量监测或检验项目的结果互认与统筹分析利用提供了技术支撑。因此,基于欧盟经验,可在资格认证的基础上,制定药品抽检质量管理体系指导原则,逐步提高抽检检验质量管理体系的针对性,促进基层药品检验机构整体专业水平进一步提升。
风险处置:建立单点联系工作组,有助于形成执法标准一致的假劣药品管控体系依托《药品质量安全犯罪与假劣药品处置合作规范》与《打击药品犯罪协定》,欧盟建立了覆盖全境的假劣药品风险处置体系。每个成员国的处置单元,通过WGEO 进行综合协调,并以SPOCs的模式进行沟通合作,联合执法并制定风险控制措施。在处置单元内部,由药品管理部门、警察执法部门、司法机关组成的SPOCs,定期组织联席会议,研讨假劣药品协同处置方式。这样“由点及面”的工作模式,促使各成员国形成一致的执法标准,对境内假劣药品进行有效管控。建议参照欧盟模式,从法规政策入手,统一风险处置标准,以点对点的方式建立由药品注册、标准管理、稽查执法、公安司法等组成的风险处置工作组,从而进一步提高风险信号挖掘与控制能力。
转化应用:多元化的样品来源途径与国际化的检验协作体系,有利于充分监测供应链体系欧盟主要进行了两方面专项研究:一是市场监测研究,旨在比较同一品种在不同成员国之间的质量差异,进而为标准提升提供建议;二是潜在假劣药品市场监测研究,主要根据问题线索评估供应链是否存在假劣药品风险。在样品来源方面,专项研究不光从市场直接抽取样品,还接受医务工作者、患者与执法人员寄送的样品,针对性地扩大了监测范围;在承检机构方面,积极与欧盟外的检验实验室合作研究。因此,建议我国在探索国抽与省抽统筹新模式的过程中,结合监管实际积极扩大样品来源,比如对常见的网售药品进行抽检;并且在设置抽检专项时,进一步拓展国际合作,以充分评估海外供应链质量安全。