您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2023-06-13 12:52
28天重复剂量吸入毒性试验
28-Day Inhalation Toxicity Study
(征求意见稿)
1、范围
本规范规定了啮齿类动物28天重复剂量吸入毒性试验的基本原则、要求和方法。
本规范适用于化妆品原料的28天重复剂量吸入毒性的评价。
2、试验目的
对于有短期反复吸入暴露可能的化妆品原料,需进行28天重复剂量吸入毒性试验。通过该试验不仅可获得一定时期内反复吸入受试物后引起的健康效应、受试物作用靶器官和受试物体内蓄积能力资料,并可估计吸入的无有害作用水平,后者可用于人群吸入的定量风险评估。
3、术语与定义
3.1 28天重复剂量吸入毒性
是指在有限时间(28天)内,每日反复通过吸入途径接触受试物后所引起的不良反应。
3.2 气溶胶aerosol
悬浮在空气中具有可以忽略下降速度/沉降速度的固态、液态或固液态混合颗粒状物质。
3.3 空气动力学直径aeodynamic equivalent diameter(AD)
气溶胶颗粒与不同直径标准单位密度(1.0 g/cm3)球形颗粒中某一颗粒的终端沉降速度相同时,该标准球形颗粒的直径即为空气动力学直径。其计量单位为μm。
3.4 空气动力学质量中位数直径mass median aerodynamic diameter(MMAD)
气溶胶中小于和等于某一空气动力学直径的颗粒总质量,占全部颗粒物质量50%时,该直径即为空气动力学质量中位数直径。
3.5 几何标准差geometric standard deviation(GSD)
用以描述气溶胶颗粒大小的分布状态。以撞击器各级累积百分比为Y轴、粒径为X轴拟合对数概率曲线,以累积百分比84.1%对应的粒径除以累积百分比50%的粒径,即为几何标准差。
3.6 理论浓度nominal concentration
制备气溶胶消耗受试物的量除以通过暴露系统的空气总体积。
3.7 实际浓度analytical concentration
暴露系统中动物呼吸区经采样分析获得的气溶胶浓度。
3.8 口鼻暴露nose-only
即仅头部、鼻部、鼻口部暴露的暴露模式,试验过程中将动物置于相应固定器内进行吸入暴露。
3.9 全身暴露whole-body
将动物置于封闭的染毒柜或腔体内进行吸入暴露的暴露模式。
4、试验的基本原则
以不同剂量受试物每日吸入给予各组实验动物,连续染毒28天,每组采用一个吸入剂量染毒。染毒期间每日观察动物的毒性反应。在染毒期间死亡的动物要进行称重和尸检。染毒结束后,处死存活动物,进行尸检、临床检查、支气管肺泡灌洗检查以及适当的病理组织学检查,以评估受试物产生的吸入毒性作用。
5、试验方法
5.1 受试物
受试物可分为气态受试物、沸点较低易挥发的液态受试物、高沸点不易挥发液态受试物、粉状或固体受试物。试验时将受试物制备成何种物态(气溶胶、粉尘)取决于受试物的理化性质、设定的浓度和/或按照实际加工使用期间的物理状态等。吸湿性的及会发生化学反应的受试物,在试验中需保证所用空气干燥,某些受试物经超声雾化时会产热,应注意选择适合的发生装置。
5.2 实验动物和饲养环境
5.2.1 动物种系的选择
常规选择啮齿类动物,首选大鼠。一般选用7周~9周龄的成年大鼠。每个性别动物体重的变动范围不应超出动物平均体重的20%。实验动物应符合国家相应规定。
5.2.2 动物的性别和数量
每一剂量组及阴性(溶媒)对照组实验动物至少应有10只(雌雄各半)。可另设追踪观察组,给予最高剂量受试物或阴性(溶媒)对照品,每组至少10只动物(雌雄各半)。
考虑到重复剂量吸入毒性试验的重要性,应适当增加每组动物数。若计划在试验过程中处死动物观察毒性反应,应增加计划处死的动物数。若考虑在试验期间增加影响动物健康的试验操作(如毒代动力学研究),则应另设卫星组,增加相应动物数仅用于满足这部分的研究需要,并不进行毒理学评价。试验结束时的动物数需达到能够有效评价受试物毒性作用的数量。追踪观察组动物在全程染毒结束后继续观察一段时间(一般不少于14天),以了解毒性作用的持续性、可逆性或迟发毒作用。
5.2.3 试验前动物准备
染毒开始前至少要有5天时间使实验动物适应实验室饲养环境。如动物拟采用口鼻暴露方式染毒,动物还需提前适应口鼻暴露时所用的固定器。
5.2.4 饲养环境
实验动物房应符合国家相应规定,选用标准配合饲料。实验饲养室内温度应控制在20℃~25℃,相对湿度在应控制在40%~70%范围内。采用人工照明,每12h明暗交替。试验期间,动物常规进食,自由饮水。当动物吸入染毒时间超过6h,则需在染毒期间以适合方式给动物喂食,并提供饮水。当动物采用全身暴露方式染毒时,每只动物在暴露期间应处在独立的空间,以防止动物聚集趴卧,其被毛吸附受试物气溶胶,影响动物吸入受试物的量。
5.3 分组
试验通常包括三个暴露水平的受试物和一个阴性/溶媒对照。阴性/溶媒对照组动物暴露于洁净空气或溶媒,溶媒应尽可能采用水,采用其他溶媒进行试验时,应根据文献报道或预试验确认所采用溶媒无吸入毒性,否则应同时设立阴性对照组和溶媒对照组,对照组的其他条件均与受试物组相同。最大吸入暴露水平的设计应在引起中毒效应的前提下又不致造成动物过多死亡,低暴露水平组应不低于人群的实际吸入暴露水平,且不出现任何毒性作用,中间暴露水平组应引起较轻的可观察到的毒性作用。
吸入试验中动物暴露水平通常以吸入暴露量表述,即气溶胶发生后经动物吸入的气溶胶的量。可根据试验周期中测得气溶胶浓度、每分钟通气量、暴露时间计算求得,公式如下:
|
吸入暴露量 |
= |
气溶胶浓度 |
× |
每分钟通气量 |
× |
暴露时间 |
÷ |
BW |
|
(mg/kg) |
(mg/L air) |
(L/min) |
(min) |
(kg) |
每分钟通气量(L/min) = 0.608×BW(kg)0.852
注:每分钟通气量可采用实际检测动物的每分钟通气量计算。也可以动物平均体重代入经验公式得出动物理论每分钟通气量。BW为动物平均体重。
5.4 吸入暴露条件
5.4.1 吸入暴露方式
首选的暴露方式为口鼻暴露,为满足特定的研究目的(如便于染毒中观察动物反应),也可采用全身暴露方式染毒,但应在试验报告中予以阐释说明。
采用口鼻暴露方式染毒时,所选用的固定器应不对动物产生额外的应激,应注意与动物体重大小匹配,并确保动物在暴露过程中无法避开吸入染毒气流。吸入装置应配备动态气流,其通风量至少应超过装置里动物总换气量的两倍,氧含量应不低于19%,并保证每只动物暴露条件稳定均一。如果进入暴露系统的空气体积小于流出体积时,应防止空气经其他途径进入暴露系统使受试物气溶胶稀释。染毒过程中应使装置内保持轻度负压,防止受试物泄露到外部环境中。
采用全身暴露方式染毒时,吸入装置需配备动态气流,每小时换气约12次~15次,应保证染毒柜内氧含量不低于19%,染毒柜内应密闭,以防止受试物泄露到外部环境中。为保证受试物气溶胶均匀分散,实验动物所占的总体积应不超过染毒柜体积的5%。
5.4.2 吸入暴露浓度
吸入染毒途径不同于其它染毒途径,其暴露水平主要通过调整气溶胶浓度,保持相同的吸入暴露时间来实现。也可调整吸入暴露时间,保持气溶胶浓度不变来实现(如采用此种方式,需排除暴露时间不一致对动物的影响)。
最大吸入浓度设计应考虑:①受试物可达到的最大浓度;②人体可能的最大暴露水平;③维持充足的氧气供应的需要;④动物福利。在缺乏限值数据支持的情况下,气溶胶最大浓度可为5 mg/L,蒸气的最大浓度可为20 mg/L,气体最大浓度可为20000 ppm。
5.4.3 吸入染毒频率
染毒期间,动物每日吸入染毒1次,每周吸入染毒7天。
5.4.4 吸入染毒时间
原则上,各组动物吸入染毒时间应一致,当采用调整吸入染毒时间来区分各组暴露水平时,各组吸入染毒时间相差不应太长,否则需要考察染毒时间不一致对动物的影响。采用口鼻暴露方式时,大鼠的吸入染毒时间应不超过6h,小鼠的吸入染毒时间应不超过4h。采用全身暴露方式时,动物的染毒时间应不少于6h,最长不超过22h。如确需超过上述时间限制,应在试验报告中予以阐释说明。
5.5 暴露条件的监测
5.5.1 浓度监测
试验期间为保证实际浓度的稳定性,可以使用实时监测设备(如气溶胶光度计),或定期通过特异性的方法(如采用撞击器吸附或化学反应的原理直接采样,然后进行化学分析),或非特异性的方法如滤膜采样法来测定呼吸区或染毒柜内气溶胶浓度,以证明暴露条件的稳定性。浓度波动范围规定:气体或挥发性受试物不得超过平均浓度±10%;液体或固体气溶胶不得超过平均值的±20%。
5.5.2 粒径监测
气溶胶颗粒的空气动力学粒径决定了在重力作用下颗粒在呼吸道中的沉降情况;气溶胶颗粒空气动力学直径的几何标准差(GSD)代表气溶胶粒度分布范围,其值越大代表粒子分布较广。为了使动物整个呼吸道充分暴露于受试物中,推荐的气溶胶颗粒空气动力学中值直径(MMAD)数值应介于1μm~3μm,GSD通常应介于1.5~3之间。可采用基于质量检测的碰撞法,基于激光飞行法的光学方法或其它替代方法定期对呼吸区或染毒柜中的气溶胶粒径进行测定,并计算GSD值。如在整个试验周期中采用替代方法进行粒径分析时,需验证替代方法所得结果与碰撞法一致。
5.5.3 暴露装置内气流
通过染毒柜或口鼻暴露系统的气流速率需严格控制,应在每次暴露期间尽可能连续监测并每小时记录1次。
5.5.4 暴露系统内环境监测
暴露系统内温度维持在22℃±3℃,口鼻暴露和全身暴露系统都需监测记录动物呼吸区的相对湿度,应在每次暴露期间尽可能连续监测并每小时记录1次。理想的相对湿度一般在30%~70%,但在某些情况下,可能无法达到(例如在测试水溶液型受试物时),应在试验报告中予以阐释说明。氧气的浓度不低于19%,二氧化碳的浓度不超过1%。
5.6 临床观察
染毒期间应每天观察,染毒结束后,追踪观察组还应继续观察至追踪观察期结束。对动物的任何毒性表现均应记录,记录内容包括发生时间、程度和持续时间。观察应至少包括如下内容:皮肤和被毛的改变、眼和粘膜变化、呼吸、循环、植物神经和中枢神经系统、肢体运动和行为活动等改变。应在首次染毒前记录动物体重,此后每周记录动物体重变化。应测定每周饲料消耗量。
5.7 临床检查
5.7.1 眼科检查
在动物染毒前和染毒后,应对所有实验动物,使用眼科镜或其他有关设备进行眼科检查。
5.7.2 血液检查
在染毒结束及追踪观察结束时应测定血球容积、血红蛋白浓度、红细胞数、白细胞总数和分类,必要时测定凝血功能如凝血时间、凝血酶原时间、凝血激酶时间或血小板数等指标。
5.7.3 临床血液生化检查
在染毒结束及追踪观察结束时进行,检查指标包括电解质平衡、碳水化合物代谢、肝、肾功能。可根据受试物作用形式选择其他特殊检查。推荐的指标包括:氯、钠、钾、禁食血糖(不同动物品系采用不同的禁食期)、血清谷丙转氨酶、血清谷草转氨酶、γ谷氨酰转肽酶、尿素氮、白蛋白、血液肌酐、总胆红素及总血清蛋白。必要时可进行脂肪、激素、酸碱平衡、正铁血红蛋白、胆碱酯酶活性的分析测定。此外,还可根据所观察到的毒性作用进行其他更大范围的临床生化检查,以便进行全面的毒性评价。
5.7.4 尿液检查
一般不需要进行,只有当怀疑存在或观察到相关毒性作用时方需进行尿液检查。
5.7.5 支气管肺泡灌洗(bronchoalveolar lavage, BAL)检查
解剖动物取肺脏,整肺称重后结扎左肺用于组织病理学检查,右肺进行BAL检查,分析BAL液的白细胞计数和分类、总蛋白、碱性磷酸酶、乳酸脱氢酶以及炎症相关细胞因子/趋化因子等参数。BAL液分析通常作为组织病理学检查的结果的补充,但不能替代组织病理学检查。
5.7.6 其他检查指标
可根据研究需要,增加额外的检测指标,如肺功能,行为学分析等。
5.8 病理检查
5.8.1 大体尸检
所有动物均应进行全面的大体尸检,内容包括动物的外观、所有孔道,胸腔、腹腔及其内容物。肺、肝、肾、肾上腺、睾丸、附睾、子宫、卵巢、胸腺、脾、脑和心脏应在分离后尽快称重以防水分丢失。应将下列组织和器官保存在固定液中,以备日后进行病理组织学检查:所有大体解剖呈现异常的器官、脑(包括延髓/脑桥、小脑和大脑皮层、脑垂体)、甲状腺/甲状旁腺、胸腺、左肺/气管、喉、鼻咽组织、心脏、主动脉、唾液腺、肝、脾、肾、肾上腺、胰、性腺、子宫、生殖附属器官*、皮肤*、食管、胃、十二指肠、空肠、回肠、盲肠、结肠、直肠、膀胱、前列腺、有代表性的淋巴结、雌性乳腺*、大腿肌肉*、周围神经、胸骨(包括骨髓)、眼及视神经、股骨(包括关节面)*、脊髓(包括颈部、胸部、腰部)*和泪腺*。
*只有当毒性作用提示或作为被研究的靶器官时才需要检查这些器官。
5.8.2 病理组织学检查
应对下述器官和组织进行检查:
(1)所有最高剂量组和对照组动物的重要的和可能受到损伤的器官或组织,如高剂量组动物的器官或组织有病理组织学的病变,则应扩展至其他剂量组的相应的器官和组织。
(2)各剂量组大体解剖见有异常的器官或组织。
(3)其他剂量组动物的靶器官。
(4)在追踪观察组,应对那些在染毒组呈现毒性作用的组织和器官进行检查。
6、试验结果的评价
6.1 数据处理
可通过表格形式总结试验结果,显示试验开始时各组动物数、出现损伤的动物数、损伤的类型和每种损伤的动物百分比。对所有数据应采用适当的统计学方法进行评价,统计学方法应在试验设计时确定。
6.2 结果评价
28天重复剂量吸入毒性试验结果应结合前期试验结果,并考虑到毒性效应指标和尸检及病理组织学检查结果进行综合评价。毒性评价应包括受试物吸入暴露剂量与是否出现毒性反应、毒性反应的发生率及其程度之间的关系。这些反应包括行为或临床异常、肉眼可见的损伤、靶器官、体重变化情况、死亡效应以及其他一般或特殊的毒性作用。在综合分析的基础上得出重复剂量吸入毒性的LOAEL和(或)NOAEL,为人群吸入的定量风险评估提供依据。
7、试验结果的解释
28天重复剂量吸入毒性试验能够提供受试物短期经呼吸系统反复吸入接触引起的毒性作用资料,也可为90天重复剂量吸入毒性试验中受试物的剂量选择提供依据。其试验结果可在有限的程度上外推到人,但它可为确定人群的允许接触水平提供有用的信息。
28天重复剂量吸入毒性试验
起草说明
为加强化妆品的监督管理,进一步提高化妆品使用安全性,中国食品药品检定研究院组织开展了28天重复剂量吸入毒性试验方法的研究制定工作。现就工作有关情况说明如下:
一、起草原则
本方法主要参照OECD 在2018 年发布的“化学品检测指南——28天(亚急性)吸入毒性研究”(TG412)编制。写作结构框架按现行《化妆品安全技术规范》进行调整。本标准制定在充分考虑动物福利的前提下,本着科学性、合理性、规范性的指导原则,对某些内容及吸入相关特征指标进行具体化和明确化,同时也尽可能提供多种吸入条件监测方法,突出体现适用性与可操作性。
二、起草过程
本研究于2021年11月由化妆品标准专家委员会立项;在研究和分析国内外相关标准的基础上,起草了本标准文本及编制说明,形成方法草案。
2023年5月本标准草案通过结题专家论证,形成如下意见及结论:按28天重复剂量吸入毒性试验和90天重复剂量吸入毒性试验分别提交方法文本和起草说明,规范方法文本,修改后通过。修改后对外征求意见,根据反馈情况对文本进行修改。
三、与我国已有相关标准的关系
我国现已发布国家标准GB/T 21754-2008《化学品28天/14天重复剂量吸入毒性试验方法》和GB/T 15670.12-2017《农药登记毒理学试验方法第12部分:短期重复吸入染毒(28天)毒性试验》。本方法在参考上述我国已有相关标准的基础上,主要针对化妆品原料的特点,以现行《化妆品安全技术规范》中亚慢性经口毒性试验和亚慢性经皮毒性试验的写作框架为模板,本着化妆品原料检测适用性及可操作性的原则进行编制。
四、与国际同类标准的关系
本方法以OECD 2018年发布“化学品检测指南——28天(亚急性)吸入毒性研究”(OECD GUIDELINES ON THE TESTING OF CHEMICALS: 28-Day (Subacute) Inhalation Toxicity Study)为依据进行撰写,结合国内实验条件及操作的具体情况,撰写的标准内容基本涵盖了实验对象、实验操作和结果分析的技术要求,重点突出了对的吸入暴露参数的规定和确认。
五、与《规范》中原方法的对比情况
现行《化妆品安全技术规范》中尚无28天重复剂量吸入毒性试验的相关内容,但有亚慢性经口毒性试验和亚慢性经皮毒性试验的相关内容,本标准以上述试验方法的写作框架为模板,重点增加了对吸入相关特征指标规定。
90天重复剂量吸入毒性试验
90-Day Inhalation Toxicity Study
(征求意见稿)
1、范围
本规范规定了啮齿类动物90天重复剂量吸入毒性试验的基本原则、要求和方法。
本规范适用于化妆品原料的90天重复剂量吸入毒性的评价。
2、试验目的
对于有长期反复吸入暴露可能的化妆品原料,需进行90天重复剂量吸入毒性试验。通过该试验不仅可获得一定时期内反复吸入受试物后引起的健康效应、受试物作用靶器官和受试物体内蓄积能力资料,并可估计吸入的无有害作用水平,后者可用于人群吸入的定量风险评估。
3、术语与定义
3.1 90天重复剂量吸入毒性
是指在实验动物部分生存期内,每日反复通过吸入途径接触受试物后所引起的不良反应。
3.2 气溶胶aerosol
悬浮在空气中具有可以忽略下降速度/沉降速度的固态、液态或固液态混合颗粒状物质。
3.3 空气动力学直径aeodynamic equivalent diameter(AD)
气溶胶颗粒与不同直径标准单位密度(1.0 g/cm3)球形颗粒中某一颗粒的终端沉降速度相同时,该标准球形颗粒的直径即为空气动力学直径。其计量单位为μm。
3.4 空气动力学质量中位数直径mass median aerodynamic diameter(MMAD)
气溶胶中小于和等于某一空气动力学直径的颗粒总质量,占全部颗粒物质量50%时,该直径即为空气动力学质量中位数直径。
3.5 几何标准差geometric standard deviation(GSD)
用以描述气溶胶颗粒大小的分布状态。以撞击器各级累积百分比为Y轴、粒径为X轴拟合对数概率曲线,以累积百分比84.1%对应的粒径除以累积百分比50%的粒径,即为几何标准差。
3.6 理论浓度nominal concentration
制备气溶胶消耗受试物的量除以通过暴露系统的空气总体积。
3.7 实际浓度analytical concentration
暴露系统中动物呼吸区经采样分析获得的气溶胶浓度。
3.8 口鼻暴露nose-only
即仅头部、鼻部、鼻口部暴露的暴露模式,试验过程中将动物置于相应固定器内进行吸入暴露。
3.9 全身暴露whole-body
将动物置于封闭的染毒柜或腔体内进行吸入暴露的暴露模式。
4、试验的基本原则
以不同剂量受试物每日吸入给予各组实验动物,连续染毒90天,每组采用一个吸入剂量染毒。染毒期间每日观察动物的毒性反应。在染毒期间死亡的动物要进行称重和尸检。染毒结束后,处死存活动物,进行尸检、临床检查、支气管肺泡灌洗检查以及适当的病理组织学检查,以评估受试物产生的吸入毒性作用。
5、试验方法
5.1 受试物
受试物可分为气态受试物、沸点较低易挥发的液态受试物、高沸点不易挥发液态受试物、粉状或固体受试物。试验时将受试物制备成何种物态(气溶胶、粉尘)取决于受试物的理化性质、设定的浓度和/或按照实际加工使用期间的物理状态等。吸湿性的及会发生化学反应的受试物,在试验中需保证所用空气干燥,某些受试物经超声雾化时会产热,应注意选择适合的发生装置。
5.2 实验动物和饲养环境
5.2.1 动物种系的选择
常规选择啮齿类动物,首选大鼠。一般选用7周~9周龄的成年大鼠。每个性别动物体重的变动范围不应超出动物平均体重的20%。实验动物应符合国家相应规定。
5.2.2 动物的性别和数量
每一剂量组及阴性(溶媒)对照组实验动物至少应有20只(雌雄各半)。可另设追踪观察组,给予最高剂量受试物或阴性(溶媒)对照品,每组至少10只动物(雌雄各半)。
考虑到重复剂量吸入毒性试验的重要性,应适当增加每组动物数。若计划在试验过程中处死动物观察毒性反应,应增加计划处死的动物数。若考虑在试验期间增加影响动物健康的试验操作(如毒代动力学研究),则应另设卫星组,增加相应动物数仅用于满足这部分的研究需要,并不进行毒理学评价。试验结束时的动物数需达到能够有效评价受试物毒性作用的数量。追踪观察组动物在全程染毒结束后继续观察一段时间(一般不少于28天),以了解毒性作用的持续性、可逆性或迟发毒作用。
5.2.3 试验前动物准备
染毒开始前至少要有5天时间使实验动物适应实验室饲养环境。如动物拟采用口鼻暴露方式染毒,动物还需提前适应口鼻暴露时所用的固定器。
5.2.4 饲养环境
实验动物房应符合国家相应规定,选用标准配合饲料。实验饲养室内温度应控制在20℃~25℃,相对湿度在应控制在40%~70%范围内。采用人工照明,每12h明暗交替。试验期间,动物常规进食,自由饮水。当动物吸入染毒时间超过6h,则需在染毒期间以适合方式给动物喂食,并提供饮水。当动物采用全身暴露方式染毒时,每只动物在暴露期间应处在独立的空间,以防止动物聚集趴卧,其被毛吸附受试物气溶胶,影响动物吸入受试物的量。
5.3 分组
试验通常包括三个暴露水平的受试物和一个阴性/溶媒对照。阴性/溶媒对照组动物暴露于洁净空气或溶媒,溶媒应尽可能采用水,采用其他溶媒进行试验时,应根据文献报道或预试验确认所采用溶媒无吸入毒性,否则应同时设立阴性对照组和溶媒对照组,对照组的其他条件均与受试物组相同。最大吸入暴露水平的设计应在引起中毒效应的前提下又不致造成动物过多死亡,低暴露水平组应不低于人群的实际吸入暴露水平,且不出现任何毒性作用,中间暴露水平组应引起较轻的可观察到的毒性作用。
吸入试验中动物暴露水平通常以吸入暴露量表述,即气溶胶发生后经动物吸入的气溶胶的量。可根据试验周期中测得气溶胶浓度、每分钟通气量、暴露时间计算求得,公式如下:
|
吸入暴露量 |
= |
气溶胶浓度 |
× |
每分钟通气量 |
× |
暴露时间 |
÷ |
BW |
|
(mg/kg) |
(mg/L air) |
(L/min) |
(min) |
(kg) |
每分钟通气量(L/min) = 0.608×BW(kg)0.852
注:每分钟通气量可采用实际检测动物的每分钟通气量计算。也可以动物平均体重代入经验公式得出动物理论每分钟通气量。BW为动物平均体重。
5.4 吸入暴露条件
5.4.1 吸入暴露方式
首选的暴露方式为口鼻暴露,为满足特定的研究目的(如便于染毒中观察动物反应),也可采用全身暴露方式染毒,但应在试验报告中予以阐释说明。
采用口鼻暴露方式染毒时,所选用的固定器应不对动物产生额外的应激,应注意与动物体重大小匹配,并确保动物在暴露过程中无法避开吸入染毒气流。吸入装置应配备动态气流,其通风量至少应超过装置里动物总换气量的两倍,氧含量应不低于19%,并保证每只动物暴露条件稳定均一。如果进入暴露系统的空气体积小于流出体积时,应防止空气经其他途径进入暴露系统使受试物气溶胶稀释。染毒过程中应使装置内保持轻度负压,防止受试物泄露到外部环境中。
采用全身暴露方式染毒时,吸入装置需配备动态气流,每小时换气约12次~15次,应保证染毒柜内氧含量不低于19%,染毒柜内应密闭,以防止受试物泄露到外部环境中。为保证受试物气溶胶均匀分散,实验动物所占的总体积应不超过染毒柜体积的5%。
5.4.2 吸入暴露浓度
吸入染毒途径不同于其它染毒途径,其暴露水平主要通过调整气溶胶浓度,保持相同的吸入暴露时间来实现。也可调整吸入暴露时间,保持气溶胶浓度不变来实现(如采用此种方式,需排除暴露时间不一致对动物的影响)。
最大吸入浓度设计应考虑:①受试物可达到的最大浓度;②人体可能的最大暴露水平;③维持充足的氧气供应的需要;④动物福利。在缺乏限值数据支持的情况下,气溶胶最大浓度可为5 mg/L,蒸气的最大浓度可为20 mg/L,气体最大浓度可为20000 ppm。
5.4.3 吸入染毒频率
染毒期间,动物每日吸入染毒1次,每周吸入染毒7天。
5.4.4 吸入染毒时间
原则上,各组动物吸入染毒时间应一致,当采用调整吸入染毒时间来区分各组暴露水平时,各组吸入染毒时间相差不应太长,否则需要考察染毒时间不一致对动物的影响。采用口鼻暴露方式时,大鼠的吸入染毒时间应不超过6h,小鼠的吸入染毒时间应不超过4h。采用全身暴露方式时,动物的染毒时间应不少于6h,最长不超过22h。如确需超过上述时间限制,应在试验报告中予以阐释说明。
5.5 暴露条件的监测
5.5.1 浓度监测
试验期间为保证实际浓度的稳定性,可以使用实时监测设备(如气溶胶光度计),或定期通过特异性的方法(如采用撞击器吸附或化学反应的原理直接采样,然后进行化学分析),或非特异性的方法如滤膜采样法来测定呼吸区或染毒柜内气溶胶浓度,以证明暴露条件的稳定性。浓度波动范围规定:气体或挥发性受试物不得超过平均浓度±10%;液体或固体气溶胶不得超过平均值的±20%。
5.5.2 粒径监测
气溶胶颗粒的空气动力学粒径决定了在重力作用下颗粒在呼吸道中的沉降情况;气溶胶颗粒空气动力学直径的几何标准差(GSD)代表气溶胶粒度分布范围,其值越大代表粒子分布较广。为了使动物整个呼吸道充分暴露于受试物中,推荐的气溶胶颗粒空气动力学中值直径(MMAD)数值应介于1μm~3μm,GSD通常应介于1.5~3之间。可采用基于质量检测的碰撞法,基于激光飞行法的光学方法或其它替代方法定期对呼吸区或染毒柜中的气溶胶粒径进行测定,并计算GSD值。如在整个试验周期中采用替代方法进行粒径分析时,需验证替代方法所得结果与碰撞法一致。
5.5.3 暴露装置内气流
通过染毒柜或口鼻暴露系统的气流速率需严格控制,应在每次暴露期间尽可能连续监测并每小时记录1次。
5.5.4 暴露系统内环境监测
暴露系统内温度维持在22℃±3℃,口鼻暴露和全身暴露系统都需监测记录动物呼吸区的相对湿度,应在每次暴露期间尽可能连续监测并每小时记录1次。理想的相对湿度一般在30%~70%,但在某些情况下,可能无法达到(例如在测试水溶液型受试物时),应在试验报告中予以阐释说明。氧气的浓度不低于19%,二氧化碳的浓度不超过1%。
5.6 临床观察
染毒期间应每天观察,染毒结束后,追踪观察组还应继续观察至追踪观察期结束。对动物的任何毒性表现均应记录,记录内容包括发生时间、程度和持续时间。观察应至少包括如下内容:皮肤和被毛的改变、眼和粘膜变化、呼吸、循环、植物神经和中枢神经系统、肢体运动和行为活动等改变。应在首次染毒前记录动物体重,此后每周记录动物体重变化。应测定每周饲料消耗量。
5.7 临床检查
5.7.1 眼科检查
在动物染毒前和染毒后,应对所有实验动物,使用眼科镜或其他有关设备进行眼科检查。
5.7.2 血液检查
在染毒结束及追踪观察结束时应测定血球容积、血红蛋白浓度、红细胞数、白细胞总数和分类,必要时测定凝血功能如凝血时间、凝血酶原时间、凝血激酶时间或血小板数等指标。
5.7.3 临床血液生化检查
在染毒结束及追踪观察结束时进行,检查指标包括电解质平衡、碳水化合物代谢、肝、肾功能。可根据受试物作用形式选择其他特殊检查。推荐的指标包括:氯、钠、钾、禁食血糖(不同动物品系采用不同的禁食期)、血清谷丙转氨酶、血清谷草转氨酶、γ谷氨酰转肽酶、尿素氮、白蛋白、血液肌酐、总胆红素及总血清蛋白。必要时可进行脂肪、激素、酸碱平衡、正铁血红蛋白、胆碱酯酶活性的分析测定。此外,还可根据所观察到的毒性作用进行其他更大范围的临床生化检查,以便进行全面的毒性评价。
5.7.4 尿液检查
一般不需要进行,只有当怀疑存在或观察到相关毒性作用时方需进行尿液检查。
5.7.5 支气管肺泡灌洗(bronchoalveolar lavage, BAL)检查
解剖动物取肺脏,整肺称重后结扎左肺用于组织病理学检查,右肺进行BAL检查,分析BAL液的白细胞计数和分类、总蛋白、碱性磷酸酶、乳酸脱氢酶以及炎症相关细胞因子/趋化因子等参数。BAL液分析通常作为组织病理学检查的结果的补充,但不能替代组织病理学检查。
5.7.6 其他检查指标
可根据研究需要,增加额外的检测指标,如肺功能,行为学分析等。
5.8 病理检查
5.8.1 大体尸检
所有动物均应进行全面的大体尸检,内容包括动物的外观、所有孔道,胸腔、腹腔及其内容物。肺、肝、肾、肾上腺、睾丸、附睾、子宫、卵巢、胸腺、脾、脑和心脏应在分离后尽快称重以防水分丢失。应将下列组织和器官保存在固定液中,以备日后进行病理组织学检查:所有大体解剖呈现异常的器官、脑(包括延髓/脑桥、小脑和大脑皮层、脑垂体)、甲状腺/甲状旁腺、胸腺、左肺/气管、喉、鼻咽组织、心脏、主动脉、唾液腺、肝、脾、肾、肾上腺、胰、性腺、子宫、生殖附属器官*、皮肤*、食管、胃、十二指肠、空肠、回肠、盲肠、结肠、直肠、膀胱、前列腺、有代表性的淋巴结、雌性乳腺*、大腿肌肉*、周围神经、胸骨(包括骨髓)、眼及视神经、股骨(包括关节面)*、脊髓(包括颈部、胸部、腰部)*和泪腺*。
*只有当毒性作用提示或作为被研究的靶器官时才需要检查这些器官。
5.8.2 病理组织学检查
应对下述器官和组织进行检查:
(1)所有最高剂量组和对照组动物的重要的和可能受到损伤的器官或组织,如高剂量组动物的器官或组织有病理组织学的病变,则应扩展至其他剂量组的相应的器官和组织。
(2)各剂量组大体解剖见有异常的器官或组织。
(3)其他剂量组动物的靶器官。
(4)在追踪观察组,应对那些在染毒组呈现毒性作用的组织和器官进行检查。
6、试验结果的评价
6.1 数据处理
可通过表格形式总结试验结果,显示试验开始时各组动物数、出现损伤的动物数、损伤的类型和每种损伤的动物百分比。对所有数据应采用适当的统计学方法进行评价,统计学方法应在试验设计时确定。
6.2 结果评价
90天重复剂量吸入毒性试验结果应结合前期试验结果,并考虑到毒性效应指标和尸检及病理组织学检查结果进行综合评价。毒性评价应包括受试物吸入暴露剂量与是否出现毒性反应、毒性反应的发生率及其程度之间的关系。这些反应包括行为或临床异常、肉眼可见的损伤、靶器官、体重变化情况、死亡效应以及其他一般或特殊的毒性作用。在综合分析的基础上得出重复剂量吸入毒性的LOAEL和(或)NOAEL,为人群吸入的定量风险评估提供依据。
7、试验结果的解释
90天重复剂量吸入毒性试验能够提供受试物长期经呼吸系统反复吸入接触引起的毒性作用资料。其试验结果可在有限的程度上外推到人,但它可为确定人群的允许接触水平提供有用的信息。
90天重复剂量吸入毒性试验
起草说明
为加强化妆品的监督管理,进一步提高化妆品使用安全性,中国食品药品检定研究院组织开展了90天重复剂量吸入毒性试验方法的研究制定工作。现就工作有关情况说明如下:
一、起草原则
本方法主要参照OECD 在2018 年发布的“化学品检测指南——90天(亚慢性)吸入毒性研究”(TG413)编制。写作结构框架按现行《化妆品安全技术规范》进行调整。本标准制定在充分考虑动物福利的前提下,本着科学性、合理性、规范性的指导原则,对某些内容及吸入相关特征指标进行具体化和明确化,同时也尽可能提供多种吸入条件监测方法,突出体现适用性与可操作性。
二、起草过程
本研究于2021年11月由化妆品标准专家委员会立项;在研究和分析国内外相关标准的基础上,起草了本标准文本及编制说明,形成方法草案。
2023年5月本标准草案通过结题专家论证,形成如下意见及结论:按28天重复剂量吸入毒性试验和90天重复剂量吸入毒性试验分别提交方法文本和起草说明,规范方法文本,修改后通过。修改后对外征求意见,根据反馈情况对文本进行修改。
三、与我国已有相关标准的关系
我国现已发布国家标准GB/T21765-2008《化学品亚慢性吸入毒性试验方法》。本方法在参考上述我国已有相关标准的基础上,主要针对化妆品原料的特点,以现行《化妆品安全技术规范》中亚慢性经口毒性试验和亚慢性经皮毒性试验的写作框架为模板,本着化妆品原料检测适用性及可操作性的原则进行编制。
四、与国际同类标准的关系
本方法以OECD 2018年发布“化学品检测指南——90天(亚慢性)吸入毒性研究”(OECD GUIDELINES ON THE TESTING OF CHEMICALS: 90-Day (Subchronic) Inhalation Toxicity Study)为依据进行撰写,结合国内实验条件及操作的具体情况,撰写的标准内容基本涵盖了实验对象、实验操作和结果分析的技术要求,重点突出了对的吸入暴露参数的规定和确认。
五、与《规范》中原方法的对比情况
现行《化妆品安全技术规范》中尚无90天重复剂量吸入毒性试验的相关内容,但有亚慢性经口毒性试验和亚慢性经皮毒性试验的相关内容,本标准以上述试验方法的写作框架为模板,重点增加了对吸入相关特征指标规定。
扩展一代生殖发育毒性试验
Extended one-generation reproductive toxicity study
(征求意见稿)
|
观察项目 |
检测指标 |
|
笼内或开放场地观察 |
姿势、阵挛、强直、眼睑闭合、竖毛、流涎、流泪、发声、犬坐、步态异常、唤醒反射、刻板症、特异行为、被毛污秽、呼吸异常。 |
|
自主控制 |
易于挣脱、易于保定、肌张力、趋向反射、触感反射、听觉反射、夹尾反射、反正反射、着地姿势、前肢握力、后肢握力。 |
|
生理指标 |
体温、体重、瞳孔反射、瞳孔大小。 |


|
序列编号 |
触发指标 |
触发条件 |
|---|---|---|
|
序列1Ba |
亲代生育力改变(着床数、妊娠率、妊娠期长短) |
生殖系统的组织病理学结果并未出现生物学相关或剂量相关的改变。 |
|
F1动情周期改变 |
虽然出现生物学相关或剂量相关的动情周期改变但未见严重的母体毒性。b |
|
|
F1窝大小的降低 |
在缺乏母体毒性或母体致死的情况下,出现了生物学相关或剂量相关的窝大小降低。b |
|
|
F1发育节点的改变(AGD、乳头数目、进入青春期、PPS、VO) |
在无体重变化介导的情况下,左侧参数出现生物学相关或剂量相关效应。 |
|
|
产后F1幼仔存活率降低 |
在缺乏严重母体毒性的情况下,出现左侧参数的变化。b |
|
|
F1幼仔出现畸形 |
在缺乏严重母体毒性的情况下,出现左侧参数的变化。b |
|
|
F1幼仔出生存活率降低 |
在缺乏严重母体毒性的情况下,出现左侧参数的变化。b |
|
|
F1幼仔体重降低 |
幼仔出现生物学相关或剂量相关的体重降低并不伴有母体动物体重的持续递减。 |
|
|
|
经确证具有干扰内分泌机制的活性* |
|
|
序列2A&2Bc |
|
构效关系分析或化学分类上具有神经毒性* |
|
|
经确证具有干扰内分泌机制的活性* |
|
|
|
经确证具有神经行为毒性* |
|
|
|
在成年动物或人具有功能或形态上的神经毒性* |
|
|
翻正反射 |
可致翻正反射降低* |
|
|
甲状腺脏器重量和病理 |
可致甲状腺重量降低或出现相关的病理改变* |
|
|
序列3c |
|
构效关系分析或化学分类上具有免疫毒性* |
|
|
经确证具有干扰内分泌机制的活性* |
|
|
|
淋巴器官出现脏器重量或病理变化* |
|
|
|
骨髓、脾脏、胸腺、淋巴结细胞成分改变* |
|
|
|
白细胞分类计数改变* |
|
|
|
在成年动物或人具有功能性免疫毒* |
Skin Absorption-In Vivo Method
(征求意见稿)
1、 范围
本方法规定了皮肤吸收体内试验方法的术语和定义、试验原则、试验方法、试验数据和报告。
本方法适用于化妆品原料经皮吸收的体内试验。
2、 试验目的
本方法阐述了化妆品原料经皮吸收的体内试验方法,以便为化妆品原料的毒性分级、标签标识和应用提供试验依据。本试验方法适用于单一成分的化妆品原料,非单一成分的原料若使用本方法进行评价,需要提供更多的科学依据说明其可行性。
3、 术语和定义
未吸收剂量(unabsorbed dose):染毒暴露以后从皮肤洗脱下来的剂量、附着在开放性覆盖物的剂量,还包括在染毒过程中从皮肤表面蒸发的量。
吸收剂量(absorbed dose):染毒部位皮肤处理以后,包括在尿液、笼具清洗液、粪便、呼气(如果可测)、血液、各种组织(如果可采集)以及尸体残留的剂量。
可吸收剂量(absorbable dose):皮肤清洗后存留在皮肤上或皮肤内的剂量。
4、 试验的基本原则
将受试物(如放射标记物)以符合实际使用情况的制剂形式按一个或多个剂量涂布于动物剃毛后的皮肤上,然后覆盖上合适的保护装置(开放式、半封闭式和封闭式)以防止动物舔舐样品,并让受试物制剂与动物皮肤持续接触暴露一段时间。在暴露时间结束后,取下覆盖物并用合适的清洁剂清洗动物皮肤,保留用过的覆盖物和清洗液以备分析测定,然后换上新的覆盖物。在试验前、试验期间和试验结束后,动物需要一直单独饲养于代谢笼中,收集其排泄物和呼出气以备分析测定。如果有充足的证据表明极少产生或不产生挥发性的放射性代谢产物,则可以不收集动物呼出气。每个试验通常包括多个试验组,其中一个试验组动物在染毒结束后立即处死,其他试验组动物可根据试验计划在染毒结束后间隔不同时间处死。在每一个采样时间点处死相关存活动物,采集血样以及染毒部位皮肤进行分析测定;此外,对残留在动物尸体内未被排泄出的代谢物也要进行分析测定。最后,用适当的分析方法对采集的样品进行分析测定,并估计透皮吸收的程度。
5、 试验方法
5.1 受试物
受试物应该作为一个整体对其透皮吸收特性进行研究。理想情况下,应该用放射性标记受试物。
受试物应进行适当配制后用于测试。受试物的配制(比如纯品、稀释品或者含有受试物的制剂)应与人类或者其他目标种属动物接触该物质的形态一致或相似;任何不同于实际使用情形的改变均应给出合理的解释。必要时可以将样品溶解或者均匀分散于溶媒中,应该了解溶媒(水除外)的皮肤吸收特征以及与受试物的相互作用。
5.2 实验动物和饲养环境
5.2.1 动物物种、品系的选择
大鼠是最常用的实验动物,但是也可以根据检测原料性质的不同使用皮肤吸收率和人类更相似的裸鼠以及其他品系动物。一般选用成年健康的单性别动物(通常用雄性)。试验开始时,实验动物体重差异不应大于平均体重的20%。比如,雄性大鼠体重一般应在200~250g范围之内,最好是在该范围的上半区内。
5.2.2性别和数量
每个样品采集以及每个设定的观察时间点需设一个试验组,每组至少4只相同性别动物。在每一段时间间隔处死一组动物,比如在染毒结束时(通常是6h或24h)和后续观察期结束时(例如48h和72h)。如果已有资料表明受试物的经皮毒性有明显雌雄性别差异,应选用更敏感的性别;如果没有资料,则任选一种。
5.2.3饲养环境
动物实验室温度应在22℃±3℃;相对湿度应在30%~70%之间,但尽可能达到50%~60%。人工控制光照,明暗12h交替。动物常规饲料喂养,自由饮水。在试验期间(最好也包括适应期间),动物应在代谢笼中单笼饲养。因食物和水的溢出会影响到试验结果,要尽量避免及减少此类情况发生。
5.3 试验步骤
5.3.1 试验动物的准备
标记动物以便识别,并在研究开始前进行至少五天适应性喂养,使动物适应实验室条件。
适应期后(大约染毒前24小时),剃除每只动物肩部和背部的毛发。因受损皮肤后可能改变皮肤的渗透特性,因此剃除毛发时要注意避免损伤皮肤。剃毛后(大约在皮肤染毒前24小时),用丙酮轻轻擦拭皮肤表面以去除皮脂。不建议另外用肥皂和水清洗皮肤,因为任何肥皂残留物都可能促皮肤对测试物质的吸收。剃毛面积应该足够大,以便能够较准确的计算出每平方厘米皮肤上受试物的吸收量。对于体重在200~250 克范围内的大鼠,剃毛皮肤面积应该至少为10 cm2。动物准备完毕后,重新放回代谢笼。
5.3.2 皮肤染毒
选用特定面积的特定皮肤位置。然后将已知量的测试制剂均匀地涂于该部位。该量通常应模拟潜在的人体用量,通常为1~5 mg/cm2 的固体或10μl/cm2的液体。可根据试验具体条件、试验目的以及受试物的物理性质等调整染毒剂量,但应有适当理由。染毒后,受试部位应避免被触碰。通常,受试部位应采用非闭合的遮盖装置覆盖(如使用透气尼龙纱布),典型装置的示例如图1所示。某些特定情况下,受试部位应采取闭合性保护。若半挥发性测试物质的挥发导致受试物的回收率超出可接受的范围(另见5.4),此时则有必要在在受试物装置上覆盖活性炭过滤器以捕获挥发的受试物(见图1)。任何覆盖物都不应损伤皮肤,也不会吸收或与受试制备物发生反应。染毒后,将动物放回单独的代谢笼中以收集排泄物。

图1 一种用于覆盖和保护受试部位皮肤的典型遮盖装置设计示例图
5.3.3 染毒时间和取样
染毒期是指在皮肤开始接触受试物,至清洗去除受试物之间的时间间隔。所采用的染毒时间(一般为6或24h)应与通常人体可能的接触时间相对应。染毒结束后,将动物饲养于代谢笼中保留直至预定的处死时间点。染毒结束后,应对受试皮肤进行观察并记录任何可见的刺激症状。
在整个研究过程中,应采用代谢笼单独收集尿液和粪便,并可在14C-标记的二氧化碳和挥发性14C化合物(>5%)产生时进行收集和定量分析。尿液、粪便和捕获液(如14C标记的二氧化碳和挥发性14C化合物)应在每个取样时间点从每组中单独收集。若有足够的信息证明很少或几乎不产生放射性代谢物,则可采用开放式笼架。整个试验期间,应定期观察动物的毒性/异常反应。
染毒期,从开始皮肤接触后至24h以及随后的每一天,均应收集排泄物直至试验结束。一般而言,虽然三个排泄物收集间隔时间点通常就足够了,但某些受试制备物的特征或现有动力学数据可能会要求有更合适的或更多的收集时间点。
染毒结束时,从每只动物身上移去保护装置并单独保留以便试验分析。所有动物的受试部位皮肤应用棉签蘸取清剂清洗至少3次。必须注意避免污染其他部位。清洗剂应为肥皂水等有代表性的常用卫生清洁剂。最后,擦干皮肤,用新的干净遮盖装置覆盖受试部位皮肤,将这些动物放回代谢笼,组成后面时间点的试验动物组。必须保留所有擦洗拭子和洗液以供研究分析。
5.3.4 试验观察和检测
对于每个实验组,应在预定时间处死试验动物,收集血样,移去保护下的装置或覆盖物并进行分析。剪下每只动物的受试部位皮肤以及对应的空白对照部位皮肤,分别进行试验分析。受试部位皮肤可分层,可将角质层与下面的真皮层分离,以提供受试物更多的信息。在染毒之后的这一时段受试物分布的试验分析,将提供其在角质层中转运的某些特征。最后清洗皮肤并处死动物后,应移去保护下的覆盖物,以便于皮肤分层处理。从受试动物身体上环状切下受试部位的皮肤并钉在板上,将粘附性胶条贴于皮肤表面,轻压,胶条将部分角质层粘附下来,继续用胶条重复粘贴,直到胶条不在粘附与皮肤表面,表明角质层已被全部去除。将同一只动物的所有胶条合并至一个单独的容器中,加入组织消化液溶解角质层。
保留每只动物的尸体以便试验分析。在对动物尸体进行吸收剂量试验前,每个可能的靶组织均可被分离单独进行试验。一般而言,仅对尸体的总含量进行分析即可,若有其他研究提示,也可将靶组织分离进行单独试验。动物处死时,膀胱中的尿液应与先前收集的尿液合并在一起进行测试。此外,收集了代谢笼中的粪便后,笼子和捕获装置均应采用合适的溶剂冲洗并收集,其他潜在的受污染仪器也应该进行类似的处理。
5.4 试验分析
所有试验均应达到足够的回收率(如平均100±10%的放射性),超出回收率范围的应说明理由。每个样品所使用的剂量应采用适宜、有效的方法进行分析。
统计分析应考虑测定每个剂量组的平行样之间的偏差。
6.数据和报告
6.1数据
6.1.1 应对每只动物在每个取样时间的受试物和(或)代谢物进行以下测定(除单个动物的数据外)以便计算吸收剂量、未吸收剂量和可吸收剂量,且不同取样时间点的数据均应按组报告其平均值:
- 保护器具上的量;
- 从皮肤上清除下来的量;
- 在皮肤上/皮肤内、不能从皮肤上洗下的量;
- 血样中的量;
- 排泄物和呼出气体中的量(如适用);
- 残留在尸体和取下单独试验的器官上的量。
6.1.2 排泄物、呼出的空气、血液和尸体中的受试物和/或代谢物的量,用以确定每个时间点的吸收总量,从而计算出染毒后每平方厘米皮肤吸收受试物的量。
6.2 报告
试验报告应符合试验方案所规定的要求,包括所使用试验系统合理性,具体包括以下内容:
1)受试物:
- 识别数据[如,CAS 编号;来源;纯度(放射化学纯度);已知杂质;批号];
- 物理性质、物理化学性质(如,pH、挥发性、溶解度、稳定性、分子量和辛醇/水分配系数的对数值lgPOW);
2)受试物的制备:
- 配方和使用理由;
- 受试物制备详细过程、使用量、达到的浓度、赋形剂、稳定性和均一性;
3)受试动物:
- 使用的种属/系;
- 动物的数量、年龄和性别;
- 动物来源、饲养环境、饲料、生产许可证明、使用许可证明等;
- 试验开始时每只动物的体重;
4)试验条件:
- 染毒的详细过程(受试部位、分析方法、闭合/非闭合、体积、提取、检测);
- 食物和水质的详细信息。
5)结果:
- 任何毒性症状;
- 吸收数据列表(以速率、数量或百分比表示);
- 试验的总体回收率;
- 结果的解释,与该受试物已有的经皮吸收的数据进行比较。
6)结果讨论。
7)结论。
皮肤吸收体内试验方法起草说明
为配合《化妆品新原料注册备案资料管理规定》的实施,有必要建立与其相适应的体内皮肤吸收试验方法及技术指导原则,为该类化妆品原料的安全性评估提供技术保障,中国食品药品检定研究院组织起草了《皮肤吸收体内试验方法》(征求意见稿)。现就起草的有关情况说明如下:
一、起草的必要性
皮肤有吸收外界物质的能力,称为经皮吸收。皮肤最主要的吸收途径是渗透入角质层细胞,再经表皮其他各层到达真皮而被吸收(主要吸收脂溶性物质);除此之外,还可通过毛囊、皮脂腺和汗腺导管而被吸收(主要吸收水溶性物质);极少量的物质如钾、钠、汞等可通过角质层细胞间隙吸收。
皮肤吸收功能对维护身体健康必不可少,并且是外用药物治疗疾病及化妆品发挥作用的理论基础,各类外用药物利用皮肤的吸收能力达到治疗疾病的作用,各种美白护肤化妆品利用皮肤的吸收能力来达到祛斑、美白等功效。
在化妆品研究领域,进行经皮肤暴露的安全评估时,皮肤吸收率的检测是一项基础而又非常重要的内容。经皮吸收试验方法可分为两类:体外吸收方法和体内吸收方法,其中,用各类实验动物开展的体内皮肤吸收试验方法能够为受试物的经皮吸收率提供较为可靠的资料。
国外现有的体内皮肤吸收评价方法或指导原则有世界卫生组织(World Health Organization, WHO)2006年发布的《环境健康标准235:皮肤吸收》(Environmental Health Criteria 235: DERMAL ABSORPTION),经济合作与发展组织(Organization for Economic Co-operation and Development,OECD)2004年发布的《皮肤吸收:体内试验方法》(OECD guideline for the testing of chemicals Skin Absorption: in vivo Method),欧洲食品安全局(European Food Safety Authority, EFSA)2017年发布的《皮肤吸收指南》(Guidance on dermal absorption),以及美国环保署(United States Environmental Protection Agency,US EPA)1998年发布的《健康效应测试指南:皮肤渗透》(Health Effects Test Guidelines OPPTS 870.7600 Dermal Penetration)等。我国于2011年发布了国家标准《化学品皮肤吸收体内实验方法》(GB/T 27825-2011),该标准所用的试验方法与OECD的方法基本一致。
2021年,我国发布了《化妆品新原料注册备案资料管理规定》,对化妆品新原料注册备案资料要求进行了明确规定,其中在国内外首次使用的具有较高生物活性的寡肽、多肽、蛋白质类新原料,以及拟用于皮肤部位的纳米新原料,要求提交皮肤吸收/透皮试验资料。因此,有必要建立与其相适应的体内皮肤吸收试验方法及技术指导原则,为该类化妆品原料的安全性评估提供技术保障。
二、起草原则与思路
查阅国内外关于药物、化学物质皮肤吸收的体内试验方法,对不同的试验方法进行对比、分析,从而转化、建立适用于化妆品原料皮肤吸收率检测的试验方法,用于具有较高生物活性物质或纳米级物质的皮肤吸收率检测。在建立体内皮肤吸收率试验方法时,明确对受试物的要求、实验动物的选择及饲养条件、实验组和对照组的设置、阳性对照物的选择、皮肤染毒方式、染毒暴露时间、样品采集与检测、数据统计方法、结果判定原则等内容,并起草皮肤吸收体内试验方法实验报告范例。
根据研究所得的试验方法,制定技术指导原则,如适用范围、评价指标、统计学方法、结果判定原则等,建议作为具有较高生物活性物质或纳米级物质等申报化妆品新原料时的安全性评价技术审评原则。
三、主要内容
(一)研究方法
查阅国内外关于药物、化学物质皮肤吸收的体内试验方法,通过对比分析,转化为化妆品用原料皮肤吸收体内试验方法,明确其适用范围、受试物要求、实验动物的选择与饲养、暴露方式及条件、样品采集与检测、数据统计方法、结果判定原则等。在此基础上,梳理该试验方法的关键技术指标,通过专家论证及征求意见,并对反馈意见进行了研究和吸纳,起草了皮肤吸收体内试验技术指导原则。
1.实验动物。根据国内外文献、动物伦理要求和试验指导原则对试验动物的种类、性别、体重、数量、饲养条件、适应性饲养时间等条件进行规定,并根据皮肤吸收试验的特点规定动物每笼饲养数量、动物分组等。
2.暴露方式及条件。根据受试物的物态对皮肤吸收的暴露方式进行规定,包括受试部位、受试物浓度及用量、受试部位的保护、暴露时间等。
3.样品采集与检测。根据受试物及实验动物的特点,对样品采集的范围及时间点进行规定,包括实验动物的尿液、粪便、捕获液、遮盖物、擦洗试子、洗液、血样、受试部位皮肤、动物尸体或靶组织、饲养笼清洗液等,并采用适宜、有效的方法进行检测。
4.数据统计分析。对试验数据的统计分析方法进行规定,包括不同时间点的回收率、吸收剂量、未吸收剂量、可吸收剂量等,从而计算出染毒后每平方厘米皮肤吸收受试物的量。
5.试验报告编写。根据所制定的试验方法对试验报告的内容进行规定,包括受试物的描述及制备、实验动物相关信息、染毒方法、试验结果的统计分析及解释、试验结论等。
6.技术指导原则制定。根据所制定的试验方法及试验报告的内容,制定相应的技术指导原则,包括该试验的适用范围、评价指标、统计学方法、结果判定原则等。
(二)技术路线

急性吸入毒性试验急性毒性分级法
Acute Inhalation Toxicity Acute Toxic Class Method
(征求意见稿)
1、 范围
本试验方法规定了急性吸入毒性试验急性毒性分级法的试验原则、试验方法、数据和报告。
本试验方法适用于化妆品原料急性吸入毒性的急性毒性分级。
2、 试验目的
本试验方法的目的是通过测试获得受试物的健康危害信息等资料,从而可参考联合国全球化学品分级和标签管理协调制度(the United Nations(UN)Globally Harmonized System (GHS)of Classification and Labelling of Chemicals)的化学品急性吸入毒性分级标准对其急性毒性进行分级。本试验方法适用于单一成分的化妆品原料,非单一成分的原料若使用本方法进行评价,需要提供更多的科学依据说明其可行性。本试验方法不适用于测试难溶的同分异构物、纤维类、纳米类原料等。
3、 定义
急性吸入毒性分级法是指按照一定的连续试验步骤和相应的浓度进行测试,从而获得受试物的毒性分级和半数致死浓度(LC50,Lethal Concentration 50)估测范围的试验方法。
理论浓度(nominal concentration),是指应用受试物的量除以通过染毒系统空气的总体积所得到的浓度。
实际浓度(actual concentration)是指在吸入式染毒柜中动物呼吸道中的受试物浓度,可通过特异性(如直接采样、吸附或化学反应等方法进行采样、分析)或非特异性(如滤膜增重法)等方法进行测试分析。
4、 试验原则
本试验采用分步试验的方法,观察试验动物段时间(一般为4 h,不超过24 h)吸入暴露后的反应,从而得到可以对受试物急性吸入毒性进行分级的信息。每步试验使用每种性别3只动物在确定的浓度下进行吸入暴露试验。根据动物死亡和/或濒死状态,有时只进行2步试验操作就可以对受试物的急性吸入毒性类别做出判断。如果有证据表明某种性别的动物比另外一种动物的敏感性要高时,可以只使用敏感性别动物进行试验。前面一步操作的试验结果决定着下一步试验操作:
a)无需进行下一步试验;
b)每种性别3只动物;或
c)只使用6只敏感性别的动物。方法是每个试验浓度组使用6只敏感性别的动物(性别不限),以估测的毒性类别界限范围数值中较低的浓度来进行试验。
应人道地处死濒临死亡的、处于难以忍受疼痛的和严重持久痛苦的动物按照试验死亡进行统计。
5、 试验方法
5.1方法描述
5.1.1 动物种属的选择
应使用健康年轻、实验室中常用的种属动物进行试验。首选大鼠,如使用其他动物应进行论证,说明理由和依据。
5.1.2 动物的准备
雌性动物应是未经产的、未妊娠。要保证每步试验暴露时的鼠龄在8周~12周;每一步试验之间使用的同周龄、同性别动物的平均体重相差应不超过±20%。采用随机选择原则,并对每只动物做出标记。在进行试验之前至少要在笼内饲养5天以适应实验室饲养条件。试验前还需在试验设备中进行短期的适应训练,以减少因为进入新的设备环境造成的应激。
5.1.3 饲养条件
实验动物暴露前和暴露结束后,室内温度应在22℃±3℃。理想情况下相对湿度在30%~70%范围内,但试验暴露期间以水为载体时很难做到。
实验动物暴露前后按性别、浓度分笼饲养,但是每笼动物的数量不能妨碍对每只实验动物的观察,并应减少由于同种相残和相互撕咬而造成实验动物信息的丢失。以口-鼻式装置暴露时,需将动物放入染毒柜内使其适应染毒柜的环境。动物在染毒柜内的固定不能过度的增加动物体力、热量和活动的负荷,动物的固定可能影响动物的体温(过热)和/或每分钟动物呼吸量。如果有资料表明这些改变没有达到可感觉到的程度,就不一定需要预先在染毒柜内进行适应操作。全身暴露气溶胶的动物暴露期间需各自相隔互不接触,防止柜体内动物的相互趴卧造成被毛对气溶胶的过滤作用。除暴露期间外,可使用常规和经过认证的实验室饮食,自由饮水。采用人工照明,每12 h明暗交替。
5.1.4 吸入染毒装置
要根据受试物的性质和试验目的来选择吸入染毒方式。首选的染毒方式是口-鼻式染毒(包括头部暴露、鼻部暴露或者口鼻部暴露模式)。当受试物是液体、固态气溶胶和能产生气溶胶的蒸气时,首选鼻式暴露方式。对于特殊的研究目的,也可采用全身暴露染毒装置以更好地满足研究需求,但应在试验报告中予以阐释说明。为了保证全身染毒时柜体内空气中受试物的稳定性,实验动物的总体积不应超过染毒柜总容积的5%。
5.2 暴露条件
5.2.1染毒浓度
5.2.1.1推荐暴露吸入时间为4 h,但需除去达到浓度稳定平衡所需时间。根据特殊试验需要也可采用其他的暴露时限,但是需在试验报告中给予相关说明。全身暴露时的动物单独饲养,防止染毒柜内其他动物的理毛行为经口摄入受试物。吸入暴露期间需要禁食。全身暴露时可提供饮水。
5.2.1.2动物吸入暴露的受试物是气体、蒸气、气溶胶或是它们的混合物。试验时将受试物制备成的何种物态(气溶胶、粉尘)取决于受试物的理化性质、设定的浓度,和/或按照实际加工使用期间的物理状态等。具有吸湿性的和会发生化学反应的受试物在试验中需保证吸入的空气是干燥的。需注意避免使用发生爆炸的浓度进行试验。
5.2.2 颗粒粒径的分布
对所有的气溶胶和可能形成气溶胶的蒸气都需测定颗粒粒径的大小。为了使整个呼吸道都能暴露受试物,推荐的气溶胶颗粒的质量中值空气动力学直径(Mass Median Aerodynamic Diameter,MMAD)为1μm~4μm,其几何标准差为1.5~3.0。需尽可能达到此标准,如技术上无法满足要求时需由专家做出评判。例如,金属烟尘比本标准的粒径要小,但是带电的粒子、纤维和吸湿性受试物(在呼吸道潮湿环境下其粒径增加)可超过这个标准。
5.2.3 载体选择与受试物的配制
为了制备具有合适浓度和粒径大小的受试气体,需要使用载体溶剂,水是首选载体溶剂。颗粒物需要机械加工达到要求的粒径大小,但要注意加工中是否引起受试物的分解和变性。如机械加工改变了受试物组成成分(由于过度摩擦产生高温),受试物的组成要经过检测分析进行确认。需注意不要污染受试物。对于不能形成可吸入性的或不易形成粉末的颗粒材料不需要进行试验。磨损试验是用来证明加工这些材料时不会产生呼吸性颗粒物。如果经磨损试验证实可产生呼吸性颗粒物,该被加工的材料应当进行吸入毒性试验。
5.2.4对照组
当使用的受试物载体及阴性(仅吸入气体)的有历史背景数据时,一般不需要平行设置载体对照组或阴性(仅吸入气体)对照组。
5.3 暴露条件的监测
5.3.1 染毒装置内气流
通过染毒柜的气流速率需严格控制,可连续在线监测记录或每次暴露期间至少每小时监测记录一次。试验所用空气中受试物浓度(或其稳定性)的监测数据是对染毒系统中各个动力学参数作用的综合结果,因此这些数据可提供控制制备动态空气设备相关参数的间接方法。在口-鼻式染毒时,当通过染毒系统的试验气体的气流动力不足时,需注意动物的在染毒装置内是否发生重复吸入。已有确定在所选定的操作条件下不会发生重复吸入的方法。氧气的浓度至少为19%;二氧化碳的浓度不超过1%。如果证实确实不能达到这个标准,需要测定并记录试验气体中氧气和二氧化碳的浓度。
5.3.2染毒装置柜体内温度和相对湿度
染毒柜内温度维持在22℃±3℃。口-鼻式和全身暴露都需监测记录动物呼吸带的相对湿度,在吸入染毒的4 h之内至少3次;吸入染毒暴露期限较短的试验可以每小时监测记录1次。理想的相对湿度需维持在30%~70%;但是有时很难达到(例如:受试物的水制备物),有时是受试物与测定方法发生干扰而无法测定。
5.3.3 受试物
5.3.3.1 理论浓度
无论何时都应尽量记录并计算染毒柜空气中的理论浓度。理论浓度是指制备产生的受试物的量除以通过染毒系统的空气总体积得到的参数。虽然理论浓度不是用来描述动物暴露特征,但是通过理论浓度与实际浓度进行比较可以得到试验系统制备效率的指征,从而可发现制备染毒空气过程中存在的问题。
5.3.3.2 实际浓度
1)实际浓度是吸入染毒柜内动物呼吸道中的受试物的浓度,可以通过特异性的方法(如吸附或化学反应)直接采样,然后进行分析;或是通过非特异性的方法(如滤膜增重法)来获得。单一成分的粉末、低挥发性液体的气溶胶才能使用增重法,同时还需要专门预试验数据的支持。对多成分的粉末气溶胶也可以用增重分析法测量,但是这需要证实所制备的空气与最初受试物的组成成分相似。如果没有此类数据资料,则需在试验期间按照规定的时间间隔对制备产生的空气进行再分析(理想的是染毒柜中的空气)。对可挥发的或易升华的气溶胶,需证实所用方法能收集到所有物体状态的受试物。需在试验报告中报告靶浓度(目标浓度)、理论浓度和实际浓度,但只有实际浓度可用来计算致死浓度值。
2)尽量使用同一批试验受试物。应将试验样本保存在能维持纯度、均匀性和稳定性的条件下。开始试验前对受试物的特征有充分的了解和描述,如纯度、鉴别特征和鉴定出来的污染物和杂质。这可用以下参数来证实,如保留时间、相对峰面积和通过质谱、气相色谱或其他的检测方法得到的分子量,包括且不限于以上内容。检验检测机构需要对委托方提供的受试物进行必要的简单描述(如颜色、物理状态等)。
3)需尽可能的保持染毒空气成分稳定,可采用适当的分析方法连续或间隔采样监测。间隔采样时,吸入染毒4 h的试验至少采样2次。如果由于系统内染毒空气流量过低所限,或染毒浓度太低需采集较多的空气样本时而不能完全满足连续或间隔采样量的要求,可在整个暴露期内只采集一次样本。如果样本之间出现明显波动,进行下一个浓度试验时需在暴露期间采集4次样本进行分析。空气各个样本之间的浓度波动范围规定,气体和挥发性受试物不得超过平均浓度±10%,液体或固体气溶胶不超过平均值的±20%。并需要计算柜内空气受试物达到95%平衡时的时间(T95)。一个暴露周期内需要考虑到制备受试物空气的时间和达到T95的时间。对组成非常复杂混合物的蒸气/气体、气溶胶(如推动终端装置设备产生的燃烧空气和试验物形成的)在染毒柜内空气中每一相成分物相各不相同,至少要选择一个标志性物质来进行分析,通常是受试制备物中处在某一物相(蒸气/气体或气溶胶)的主要的活性物质。如果试验物质是制剂(商品形式的产品)分析报告的浓度应是制剂的总浓度,不是活性成分的或某成分的浓度。
5.3.3.3 颗粒物粒径的分布
气溶胶的粒径大小需在染毒的4 h期间至少测定2次,方法是使用撞击采样器或其他替代的设备如空气动力学粒径仪等。如果撞击时采样器与替代的测定方法所得到的结果相同,在整个试验期间的就可以使用替代的方法进行监测。后一种检测方法,如滤膜增重或尘埃测定器/气体发泡收集管等,与最初确定的测定方法进行平行试验,来验证原来测定方法的收集效率。粒径大小分析得到的质量浓度应与采用滤膜分析法得到的质量浓度的一个合理的限度之内。如果在试验开始阶段就证明两种试验方法的质量浓度相同,就可以免去验证性试验。从动物福利方面考虑,需采取措施减少无确定结果的数据资料,因为这会要求重复进行暴露试验。如果空气中的蒸气受试物能发生凝结而形成气溶胶,或者染毒蒸气空气中测定到颗粒物也就是暴露空气存在潜在的混合相,需对这种受试物进行粒径的测定。
5.4 操作步骤
5.4.1正式试验(main test)
5.4.1.1 每一步使用雌雄各3只动物,或6只敏感性别的动物。如果采用鼻式暴露时使用的啮齿类动物不是大鼠,需要调整最长的暴露时间以减少动物种属之间的差异。可从4个固定的起始浓度水平染毒,这个浓度应能引起染毒动物中的某些动物出现毒性作用。气体、蒸气和气溶胶的试验步骤图(见7试验步骤流程图)的浓度水平是第1类~4类的分级标准的界限值:气体为(100、500、2500、20000 ppm/4 h)(见图1);蒸气为(0.5、2、10、20 mg/L/4 h)(见图2);气溶胶为(0.05、0.5、1、5 mg/L/4 h)(见图3)。高于各相应物态类别的上限浓度就归为第5类。按照各自的试验图表来确定起始浓度。根据人道处死和动物死亡的数量,按照图中的指示的箭头来决定下一步的试验,直到能对受试物作出毒性分级时结束试验。
5.4.1.2 进行下一个暴露浓度组试验的时间间隔长短要根据毒性体征的发作时间、持续时间和严重程度来决定。决定进行暴露下一个浓度水平需要直到前一个浓度水平的动物存活状态完全确定下来后再进行。建议的每个浓度水平之间的时间间隔为3至4天,这可以观察到可能存在的迟发毒性。如出现的反应不能确定,可适当调整时间间隔的长短。
5.4.2 限度试验(limit test)
5.4.2.1如已知或预期受试物几乎无毒时可进行限度试验,如在超过规定的极限浓度值以上时才引起毒性反应的受试物。受试物的毒性的信息可从已经检测的类似化合物、混合物或产物结果得到,此时要考虑到已知的具有毒理学意义的受试物的特性、混合物构成的百分比等。在没有或很少毒性信息的情况下,或者是估计受试物是具有毒性,需进行正式试验(可参考OECD指南39号)。
5.4.2.2 正常的操作程序下,使用每种性别3只动物,或使用6只敏感性别的动物,暴露水平分别是:气体20000 ppm;蒸气20 mg/L;粉尘/雾5 mg/L(如果技术可达到)。受试物制备成气溶胶时,粒径的大小需达到可呼吸性粒子的大小(如MMAD为1μm~4μm)。绝大部分的受试物都能在2 mg/L的浓度水平进行试验。如进行高于2 mg/L浓度水平的气溶胶试验,受试物应能够制备成可呼吸性粒子大小。从动物福利方面考虑,不主张超过限度浓度的试验。
5.4.2.3 只在有可能直接涉及到人类健康需要的情况下才进行第5类染毒浓度水平(>20000 ppm或mg/L)的试验,并需在试验报告中论述理由。对潜在爆炸性受试物试验设计时要认真研究避免引起发生爆炸的条件出现。为了避免不必要的使用动物,开始限度试验前,需在染毒系统不放置动物的条件下进行预试验,以确认染毒条件是否能达到限度试验的水平。
5.4.3 观察
5.4.3.1 暴露期间要对实验动物经常进行临床观察。暴露染毒结束后的当日至少要观察2次,还可根据动物对染毒的反应增加观察次数;此后的14天内至少每日观察1次。观察周期时长不是固定不变的,需要根据出现的临床症状的特点、时间以及恢复期的长短决定。毒性反应出现和消失的时间很重要,应特别注意是否存在毒性体征延迟的倾向,对每只动物的所有的观察都要分别作系统的记录。
5.4.3.2 对处于濒死的动物和表现出严重的疼痛和/或持续的严重痛苦的动物从动物福利考虑要人道的处死。对最初出现的微弱的毒性体征和由于暴露操作引起的短暂的呼吸道的异常改变等临床体征要认真分析区别,不要错误认为这些都是染毒处理引起的毒性作用。要按照列在人道观察终点的指南文件中“人道终点”的原则和标准进行操作。对人道处死的或发现已经死亡的动物处死和死亡时间要尽可能的准确的记录。笼外观察包括动物皮肤和被毛、眼和粘膜、呼吸、循环、自主神经和中枢神经系统的改变,以及身体活动和行为方式等变化。尽可能的注意局部作用与系统作用之间的任何差别。关注点主要为颤动、抽搐、流涎、腹泻、嗜睡、睡眠和昏迷。测定直肠温度可辨别出是否存在因染毒或在染毒柜内过度的限制造成的反射性呼吸过缓、体温过低/过高。
5.4.4 体重
进入实验室适应环境期间每只至少要称重一次;染毒当日开始试验前(第0天)、染毒后的1、3、7天(此后每周记录一次)、染毒第1天后死亡或进行人道处死的动物进行称重。体重变化是判定毒性作用的指标,并且对于体重持续低于染毒前体重的20%要给予密切观察。试验结束时存活动物称量体重后并人道地处死。
5.4.5 病理学
所有实验动物,包括试验期间死亡的、人道处死的动物和动物福利考虑从试验队列中退出的,都需进行大体解剖检查。如果发现动物死亡后不能立即进行解剖检查,将死亡动物放入冰箱的冷藏箱内低温存放,以减少动物尸体的自溶分解。大体尸检应在死亡后1 d~2 d进行。每只动物大体解剖所见的病理学改变都记录,特别注意呼吸道出现的任何变化。其他辅助的检查,包括可能对试验结果做出解释的检查指标,如测定存活大鼠的肺脏重量或者对呼吸道进行显微检查以提供受试物刺激性的证据等。检查的器官涉及到染毒后存活24 h以上的动物出现大体解剖学改变的器官、或者已知或预期可能受到影响的器官。全呼吸道的显微组织学检查为鉴定受试物是否与水发生反应(如,酸和吸湿性的受试物反应)提供非常有价值的信息。
6、 数据和试验报告
6.1 数据
需提供每只动物的体重、大体解剖检查所见的全部资料。临床观察的数据需要总结成表格的形式以体现出每个试验组使用的动物数、出现特异性毒性体征的动物数、试验期间死亡或人道处死的动物数、每只动物的死亡时间、详细描述毒性作用的发生、发展的时间进程和体征的可逆性;大体解剖所见。
6.2 试验报告
试验报告需包括以下信息:
1)实验动物和饲养条件:
— 饲养笼具的描述:每笼动物的数量(或动物变化的数量)、垫料、环境温度、相对湿度、光照周期、饲料合格认证资料;
— 使用的动物种属和品系,不使用大鼠进行试验的论证依据;
— 动物的数量、周龄和性别;
— 随机选择分组的方法;
— 动物食物、饮水的质量(包括饲料的类型/来源和供水的来源);
— 试验前的所有的包括饲料、检疫、疾病的处理等描述。
2)受试物:
— 物理性状、纯度、相关的理化性质(包括同分异构体);
— 受试物特征性数据和化学文摘(CAS)注册号(如果已知)。
3)载体:
— 如果不是用水作为载体,所用的载体是什么,对采用这种载体的理由作出论述;
— 载体的历史背景数据或所使用的载体不会干扰试验结果的数据资料。
4)吸入染毒装置:
— 吸入染毒装置的介绍,包括大小尺度和体积;
— 除了描述制备染毒空气操作外,还需动物暴露所用设备及其来源进行描述;
— 测定气温、湿度、粒径大小和实际浓度的仪器设备;
— 空气的来源、空气供给/解压处理、调节供给条件所使用的系统;
— 确保试验空气均匀所使用的调节设备和方法;
— 压差(正压或负压);
— 每个吸入装置动物暴露孔的数量(口-鼻式装置);每个染毒装置的动物数(全身染毒设备);
— 试验时空气中受试物的均匀性、稳定性;
— 温度和湿度感受器、采样孔所在的部位;
— 空气流速,包括口-鼻式固定孔处的空气流速、全身染毒柜内放置动物处的空气流速;
— 如果测定了空气中氧气和二氧化碳需要提供所用设备的信息;
— 达到吸入染毒柜空气浓度稳定平衡的时间(T95);
— 每小时空气体积更换次数;
— 计量仪器设备。
5)暴露数据:
— 正式试验中选择设定靶浓度的理由(基本原理);
— 理论浓度(制备的试验物质进入到吸入染毒设备的总量除以通过染毒柜空气的总体积);
— 从动物呼吸道中采样测得的受试物的实际浓度;对于不同物态的混合物受试物(气体、蒸气和气溶胶),需分别对每一物态进行测定;
— 报告中所有的空气浓度都用质量单位(如mg/L,mg/m3等)或体积单位(如ppm、ppb)来表示;
— 颗粒物粒径大小的分布用MMAD和GSD来表示,同时列出得到此参数数值的计算过程。应在报告中记录每个检测到的粒径大小的数据。
6)试验条件:
— 受试物制备的详细操作,包括固态物质被制备成为直径更小的颗粒物或制备成溶液的详细操作过程;如机械加工时改变了受试物的组成成分,还要提交加工后受试物组成成分的检测分析的结果;
— 制备试验空气所用设备和制备动物染毒时所用空气的操作的描述(可以用示意图表示);
— 所用化学分析方法的详细描述和分析方法可靠性的详细描述(包括空气媒介样本中受试物的回收率);
—设置染毒浓度的基本原理。
7)结果:
— 列表表示染毒装置内空气环境的温度、湿度和流速;
— 列表表示染毒空气中理论浓度、实际浓度数据;
— 列表表示颗粒粒径大小的数据,包括样本的采集测试方法、粒径大小的分布、MMAD及GSD的计算等;
— 列表表示动物出现毒性反应的数据和每只动物暴露浓度水平(如死亡率、性质、严重程度和作用的持续时间等毒性体征的动物数量);
— 试验中测得的每只动物的体重、计划试验结束人道处死前动物死亡的日期和时间、毒性体征出现的时间和过程,以及每只动物出现的毒性体征是否是可逆的等;
— 每只动物大体解剖和组织病理学检查(如果进行了组织学检查)所见;
— 受试物按照急性吸入毒性分级属于哪类和LC50界限值。
8)讨论和结果分析:
— 需要特别强调的是需要描述是否达到本标准规定的参考值范围,如限度浓度试验和颗粒粒径大小的规定;
— 应根据总体调查结果处理颗粒的可吸入性,尤其是当无法满足颗粒尺寸标准时;
— 对测定理论浓度和实际浓度所采用的方法的一致性进行评价;并对整个试验过程中都要涉及到的理论浓度与实际浓度的相互关系进行评价;
— 对动物可能的死亡原因和主要的作用机制(是系统毒性或局部毒性)作出分析估测;
— 如果因为动物疼痛、或表现出严重的持久的痛苦状态而被人道处死,要根据OECD人道终点标准的指导文件作出解释说明。
7、 实验步骤流程图
图1:各初始气体浓度应遵循的试验程序(ppm/4 h)
对于每一种起始气体浓度,本图中所列的相应检测方案概述了应遵循的试验程序。根据实施人道处死或死亡动物的数量,试验程序遵循箭头指示。
图1a:起始浓度为100 ppm
图1b:起始浓度为500 ppm
图1c:起始浓度为2500 ppm
图1d:起始浓度为20000 ppm

图1a:气体的起始浓度为100 ppm/4 h的试验程序

图1b:气体的起始浓度为500 ppm/4 h的试验程序

图1c:气体的起始浓度为2500 ppm/4 h的试验程序

图1d:气体的起始浓度为20000 ppm/4 h的试验程序
图2:蒸气的各起始浓度应遵循的程序(mg/L/4 h)
对于每一个起始气体浓度,本图中所列的相应检测方案概述了应遵循的试验程序。根据实施人道处死或死亡动物的数量,试验程序遵循箭头指示。
图2a:起始浓度为0.5 mg/L
图2b:起始浓度为2.0 mg/L
图2c:起始浓度为10 mg/L
图2d:起始浓度为20 mg/L

图2a:蒸汽的起始浓度为0.5 mg/L/4 h的试验程序

图2b:蒸汽的起始浓度为2 mg/L/4 h的试验程序
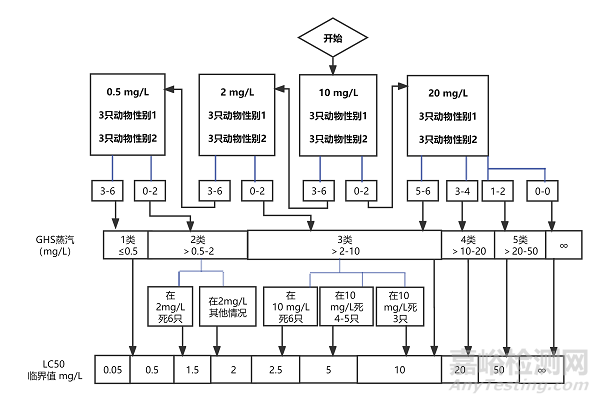
图2c:蒸汽的起始浓度为10 mg/L/4 h的试验程序
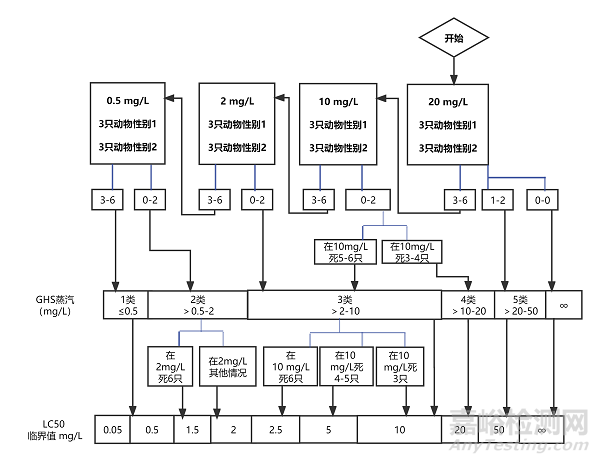
图2d:蒸汽的起始浓度为20 mg/L/4 h的试验程序
图3:粉尘和雾的各起始浓度应遵循的程序(mg/L/4 h)
对于每一个起始气体浓度,本图中所列的相应检测方案概述了应遵循的试验程序。根据实施人道处死或死亡动物的数量,试验程序遵循箭头指示。
图3a:起始浓度为0.05 mg/L
图3b:起始浓度为0.5 mg/L
图3c:起始浓度为1 mg /L
图3d:起始浓度为5 mg /L

图3a:粉尘和雾的起始浓度为0.05 mg/L/4 h的试验程序

图3b:粉尘和雾的起始浓度为0.5 mg/L/4 h的试验程序

图3c:粉尘和雾的起始浓度为1 mg/L/4 h的试验程序
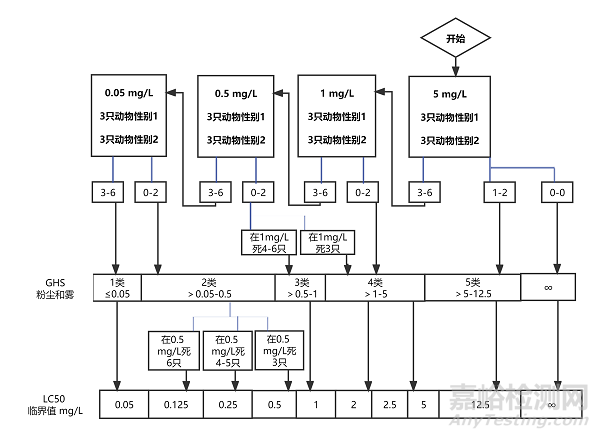
图3d:粉尘和雾的起始浓度为5 mg/L/4 h的试验程序
急性吸入毒性试验急性毒性分级法起草说明
为配合《化妆品新原料注册备案资料管理规定》的实施,有必要建立与其相适应的化妆品原料急性吸入毒性试验-急性毒性分级的试验方法,从而为化妆品原料的安全性评估提供技术保障,中国食品药品检定研究院组织起草了《急性吸入毒性试验急性毒性分级法》(征求意见稿)。现就起草的有关情况说明如下:
一、起草的必要性
随着我国在2021年2月发布《化妆品新原料注册备案资料管理规定》,针对化妆品新原料,要求提供相应的毒理学安全性评价资料、安全风险评估资料等内容。化妆品原料种类众多,接触或进入人体的方式多种多样,有经皮、经口、经过粘膜、经过呼吸道等多种方式,产生的潜在安全影响均需要全面评价,以确保化妆品原料在注册、备案的过程中的安全性得到充分的关注,从而为化妆品的安全应用奠定坚实的技术基础。
通过喷洒等方式使用的化妆品原料及产品,如具有香氛、除臭等功能的原料或产品,常常可通过吸入的方式进入体内,因此,尽快完善相应原料的安全评价技术方法,具有重要的现实意义。
国际经济合作与发展组织(Organization for Economic Co-operation and Development,OECD)基于科学技术的发展、管理规定及动物福利等方面的考虑,于1981年公布实施了急性吸入毒性测试导则(OECD 403),由于在2001年公布实施了急性经口毒性分级法(TG 423)后,才考虑编制相应的急性吸入毒性分级法(Acute toxic class, ATC)。对急性吸入毒性分级法(ATC)数据质量的回顾性评估表明该方法非常适合与对化学物的毒性分级和产品标签规定的需要。2009年9月OECD公布实施了化学品测试导则NO.436(2009)《急性吸入毒性试验:急性毒性分级法》,对急性吸入毒性的试验原则、方法、数据和试验报告、附录等内容进行了规定。2012年7月,中华人民共和国国家质量监督检验检疫总局、中国国家标准化管理委员会发布了中华人民共和国国家标准《化学品急性吸入毒性试验急性毒性分级法》GB/T 28648-2012,对化学品急性吸入毒性试验急性毒性分级法的适用范围,原则、方法、数据、报告等内容进行了规定,其中核心的主要内容是针对化学品的急性吸入毒性进行分级。
2021年,我国发布了《化妆品新原料注册备案资料管理规定》,对化妆品新原料注册备案资料要求进行了明确规定。因此,对化妆品原料的急性吸入毒性进行分级评价,对未来化妆品原料的安全状况、适用范围、使用形式等都具有重要的参考意义,值得尽快将该类技术评价方法予以完善。
二、起草原则与思路
查阅国内外关于化学品急性吸收毒性的试验方法,重点针对OECD化学品测试导则NO.436(2009)《急性吸入毒性试验:急性毒性分级法》、中华人民共和国国家标准《化学品急性吸入毒性试验急性毒性分级法》GB/T 28648-2012中规定的相关内容,对相应的试验方法进行比对、分析,进而转化,建立适用于化妆品原料安全性评价的急性吸入毒性试验急性毒性分级法。在建立急性毒性吸入毒性试验急性毒性分级试验方法时,明确了该方法适应于化妆品原料评价的适用范围,对试验目的、实验原则、实验方法中涉及的试验动物、吸入染毒装置、暴露监测、试验操作、试验观察、组织病理学结果、试验数据及报告等内容,进行了详细梳理和规定,起草了《急性吸入毒性试验急性毒性分级法》(征求意见稿)。
三、主要内容
(一)适用范围
关于《急性吸入毒性试验急性毒性分级法》适用范围的问题,通过对OECD化学品测试导则NO.436(2009)《急性吸入毒性试验:急性毒性分级法》、中华人民共和国国家标准《化学品急性吸入毒性试验急性毒性分级法》GB/T 28648-2012中相关内容的研究、分析,从试验方法的角度而言,该方法主要适用于原料急性毒性的分级,OECD和国标都是适用于“化学品”,也即是归属于化学品的原料。关于该方法在化妆品领域的适用性问题,也召开过专题讨论会,专家认为,从急性吸入毒性分级试验设置的目的来看,也只能是针对化妆品原料,只有原料才有必要对相应的急性吸入毒性进行分级,化妆品产品不存在急性吸入毒性分级的问题。因此,在该方法的适用范围方面,还是确定该方法适用于化妆品原料的急性吸入毒性分级评价,并且认为该方法适用于单一成分的原料,对非单一成分的原料,若应用该方法进行评价,需要提供更多的科学实验数据来证明其可行性。
(二)实验动物
根据国内外文献、动物伦理要求和试验指导原则对试验动物的种类、性别、周龄、体重、数量、饲养条件、适应性饲养时间等条件进行规定,并根据急性吸入毒性试验的特点对普通试验动物和敏感试验动物的性别和数量进行了特殊规定,即常规一般是每性别3只动物,或只使用敏感性别动物6只。
(三)吸入染毒途径的选择
根据试验方法对中受试物可能的吸入途径进行了规定,吸入途径包括仅头部暴露、鼻部暴露和鼻口部暴露三种吸入模式,一般首选口-鼻式染毒。
(四)暴露染毒的控制和监测
暴露条件是确保暴露染毒稳定可控的关键,涉及暴露染毒时间、受试物制备、受试物颗粒粒径分布、受试物载体配制的选择、对照动物的设置等多个方面,该方法都进行了相应的规定。
在满足暴露条件的前提下,如何对暴露情况进行监测,是确保后期染毒结果的核心,包括对染毒装置气流速率的严格控制,需连续在线监测记录或暴露期间至少每小时监测记录一次等,另外,还需要控制染毒装置的相对温度、湿度等,均是确保监测结果能符合要求的关键环节。
(五)试验操作要求
正式试验一般每步使用雌雄各3只动物,或6只敏感性别动物。根据不同的受试物状态,如气体、蒸汽或气溶胶,可根据附件中的试验步骤图,采用不同的起始浓度开展相应的试验。
(六)试验结果采集
染毒后对试验动物的观察和试验结果采集是确保试验结果科学、可靠的关键。因此,方法规定了暴露染毒后,试验观察的频率、周期、毒性体征的观察记录、动物人道主义处理的要求、试验动物的体重、组织病理学的要求等各项内容。
(七)试验报告
试验报告要求具体详实,包括实验动物、受试物、载体、吸入装置、暴露数据、试验条件、试验结果等各个方面的内容。
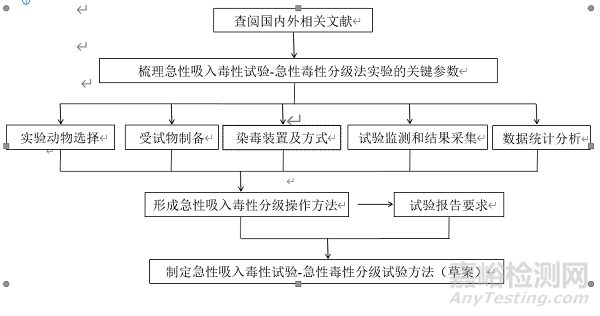
四、技术路线
体外皮肤变态反应 U937细胞激活试验
U937 cell line activation Test
(征求意见稿)
1、 范围
本方法规定了体外皮肤变态反应U937细胞激活试验的基本原则、要求和方法。
本方法适用于化妆品用化学原料潜在致敏性的评价。
2、 试验目的
本试验用于检测体外培养的人组织细胞淋巴瘤细胞表面标记物CD86的表达,以评价受试物引起皮肤变态反应的可能性。
3、 定义
3.1 70%细胞存活率浓度值 70% cell viability(CV70)
受试物染毒后,细胞存活率为70%时对应的受试物浓度值。
3.2 刺激指数 stimulation index(S.I.)
与溶剂对照相比,扣除同型对照后流式细胞仪测定的受试物阳性细胞相对强度值。
3.3 CD86有效作用浓度值 Effective Concentration 150(EC150)
CD86的相对荧光强度值达到150时对应的受试物浓度值。
4、 试验的基本原则
当致敏物质接触皮肤后,树突状细胞在移动到淋巴器官的过程中分化成熟,并上调一系列表面分子的表达。体外培养类树突状细胞:人组织细胞淋巴瘤细胞,并和受试物共暴露48h后,使用荧光抗体染料对细胞表面分子CD86染色并用流式细胞仪测定,从而评价受试物是否具有致敏性。
5、 试剂
5.1 细胞
选用人组织细胞淋巴瘤细胞(The human histiocytic lymphoma cell line,U937细胞)。细胞使用前应进行稳定性检测。推荐使用clone CRL1593.2细胞株。
5.2 培养基
RPMI 1640基础培养液中加入10%胎牛血清、适量抗生素,配制成1640完全培养基。
5.3 染色缓冲液
磷酸盐缓冲液中加入5%的胎牛血清。
5.4 染料和抗体类物质
7-氨基放线素菌-D染料(7-AAD)
FITC标记的小鼠单克隆CD86抗体(FITC-CD86)
FITC标记的小鼠IgG1(FITC- IgG1)
6、 试验方法
6.1 细胞准备
6.1.1 细胞培养
U937悬浮细胞使用1640完全培养基,于37℃、5%CO2培养箱内培养,倒置显微镜下观察细胞状态。细胞复苏一周并通过稳定性检测后可开展试验。细胞最大培养浓度不超过2×106个/ mL,复苏后使用不超过6周,传代次数不超过21代。
6.1.2 细胞稳定性检测
细胞复苏两周后,选用松香酸(AA)或2,4,6-三硝基苯磺酸作为阳性对照,乳酸(LA)作为阴性对照。当细胞可以准确对阳性物质和阴性物质进行稳定区分时视为通过稳定性检测。
6.2 受试物处理
溶剂:完全培养基(优先选择)或二甲基亚砜(DMSO)
受试物储备液:第一次运行至少选择6个浓度。于试验前一天制备,应用1640完全培养基配制0.4 mg/mL的受试物溶液,或者应用DMSO配制50 mg/mL的受试物溶液。由于没有进行剂量测定,因此在第一次运行时,应在相应的溶剂中配制6种系列浓度储备液(最终浓度为1、10、20、50、100和200µg/mL)。
受试物工作液:于试验当天制备,用完全培养基配制的受试物溶液可以直接使用,用DMSO配制的受试物溶液需用完全培养基稀释125倍得到工作溶液。将工作液等体积加入细胞悬液中进行染毒,即最终细胞接触浓度是工作液浓度再稀释2倍。
连续运行的浓度选择:基于第一次运行的结果选择至少4个第二次运行的浓度,任何后续运行的浓度都是基于之前所有运行的单个结果来选择。由于浓度间隔设置较小会影响浓度效应,所以建议以下推荐使用的最终浓度(单位:µg/mL)进行试验: 1 – 2 – 3 – 4 – 5 – 7.5 – 10 – 12.5 –15 –20 – 25 – 30 – 35 – 40 – 45 – 50 –60 – 70 – 80 – 90 – 100 – 120 – 140 – 160 – 180 – 200。
如果以上浓度无法测得受试物的70%细胞存活率浓度值(70% cell viability,CV70),可以进行浓度调整。但受试物最终细胞接触浓度最高不超过200µg /mL;如果CD86的阳性值在1ug/mL,则对0.1ug/mL进行评估,以确认不会导致CD86超过阳性阈值的受试物的浓度;DMSO最终接触浓度不得超过0.4%。
选择标准:至少2个浓度与前一次试验相同;确认非细胞毒性下的最高CD86阴性浓度;确认所有非细胞毒性CD86阳性浓度;确认最低细胞毒性浓度;在EC150(或最高阴性非细胞毒性浓度)和CV70(或溶解度评估允许的最高浓度)之间均匀选择其他所需浓度;若前两次运行有差异,尽可能选择多的共同浓度;若存在干扰,需重新选择阳性浓度。
6.3 对照物处理
培养基对照、溶剂对照(与受试物同法稀释)、阳性对照松香酸(DMSO溶解,最终浓度50µg/ml)、阴性对照LA(完全培养基溶解,最终浓度200µg/mL),每种对照需要准备三对复孔。
6.4 细胞铺板及染毒
以3 x105个/ml细胞传代培养2天或以1.5 x105个/ml传代培养3天后,调整细胞浓度为5×105个/ml,取100µL/孔受试物工作液和100µL/孔细胞悬液1:1混合接种于96孔无菌平板中,每种条件下一对复孔,一孔用于CD86染色,一孔用于IgG1同型对照染色。每次CD86表达检测均需设置培养基对照、溶剂对照、阳性对照、阴性对照各三对复孔(当使用培养基作为溶剂时,溶剂对照与培养基相同),用密封带盖住板,在37±2°C,5±1%CO2,≥95%湿度下培养孵育45±3小时。
6.5 CD86表达检测
孵育结束后,检查并确定溶解度(若在显微镜下观察到晶体或液滴,则会产生干扰)。
用排枪将细胞吹两下从96孔平板对应移至V型板中,离心弃上清;
用100µL冰冷染色缓冲液清洗一次,然后在100µL染色缓冲液中重悬细胞;
用7-AAD染色细胞(5µL/孔,10min室温,避光);
用100µL冰冷染色缓冲液清洗一次,然后在100µL染色缓冲液中重新悬浮细胞;
用FITC标记的抗CD86或小鼠IgG1(5µL/孔,30min,4°C,避光)染色细胞;
用100µL冰冷染色缓冲液清洗两次;
用100µL冰冷PBS清洗一次;
在冰冷PBS中再悬浮细胞(96孔板中100µL或流式管中200µL)。
注意:整个染色操作应在冰上进行,在每一个洗涤步骤中,不要通过反复的移液来重悬细胞,而是在弃去上清液后通过短暂的涡旋来分散细胞即可。
用流式细胞仪对每组细胞的细胞存活率及CD86阳性细胞百分比进行测定。分别以下用公式计算出受试物的细胞存活率(CV70)和受试物的细胞刺激指数(S.I.)。
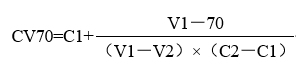
V1:大于70%最小细胞活性值
V2:小于70%最大细胞活性值
C1(μg/mL):V1对应的受试物浓度
C2(μg/mL):V2对应的受试物浓度

CD86+%:CD86阳性细胞百分比
6.6 流式细胞仪检测
绘制SSC/FSC散点图,调节仪器电压,圈出U937细胞群P1门;绘制7-AAD直方图,圈出活细胞P2门;绘制FITC/SSC散点图,放置十字门,分析细胞表面标记物CD86的表达。通过获取完全培养基/ IgG1孔表达值,设置FITC/SSC十字门分析标记,以便在可能的情况下,所有三个或至少两个完全培养基对照的IgG1位于0.6至0.9%区域内;如果这无法实现,则设置分析标记,使一个完全培养基对照的IgG1在0.6至0.9%,另一个完全培养基对照的IgG1在0.9%至1.5%;如果这仍然无法实现,则IgG1数据分布太广,必须放弃运行。之后在不移动分析标记的情况下,测量每个孔中IgG1阳性细胞或CD86阳性细胞的百分比。
细胞显示在大小(FSC)和粒度(SSC)的散点图中,设置为对数比例(Log),以便清楚地识别第一门R1门中的(population)细胞群和消除碎片。
设置流式细胞仪,每孔获得P1门细胞10000个或包括死亡细胞在内的多达20,000个细胞,或在分析开始后一分钟内获取数据。
7、 结果评价
7.1 试验成立条件
7.1.1 完全培养基对照细胞平均存活率> 90%;至少两个完全培养基对照的IgG1≥ 0.6且< 1.5%;若丢弃一个完全培养基对照的离群值后,剩下的两个完全培养基对照的校正后的CD86表达值应高于或低于其平均值的25%;完全培养基对照校正后的CD86基础表达值≥ 2%且≤ 25%。
7.1.2 DMSO溶剂对照的平均存活率> 90%;若丢弃一个DMSO对照的异常值后,剩下的两个DMSO对照的校正后的CD86表达值应高于或低于其平均值的25%;丢弃异常值后,DMSO溶剂对照CD86 S.I.的平均值小于250%。
7.1.3 阳性对照三对复孔中至少两个为阳性(CD86 S.I.≥ 150),必要时配制阳性对照系列浓度,应存在剂量反应。
7.1.4 阴性对照三对复孔中至少有两个为阴性(CD86 S.I. < 150%),且细胞活性≥ 70%。
7.2 致敏性结果判定
每种受试物至少进行两次独立的CD86表达检测试验。每种受试物表达检测试验,如果CD86 S.I.仅在最高非细胞毒性浓度下高于150%,则在第一次试验中无法判定。每次表达检测试验,如果CD86 S.I.在所有非细胞毒性浓度下低于150%,且无干扰(溶解度、颜色、细胞毒性),则该次CD86表达判定为阴性;如果CD86 S.I.高于150%,不论有无剂量反应和/或干扰,则该次CD86表达均判定为阳性。如果两次试验CD86表达结果一致,则可判定该受试物是否具有致敏性;如果两次试验CD86表达不具有一致性,则需进行第三次试验。如果第一次试验无法判定,第二次与第三次结果不一致,则需进行第四次试验。最终的预测将基于三次或四次单独运行的结果来综合判定。
7.3 有效作用浓度计算
对于判定具有致敏性的物质,进一步计算其有效作用浓度值(Effective Concentration,EC),选用以下公式计算CD86的有效作用浓度值(EC150)。

S.I.1: S.I.< 150(EC150)最低刺激指数值
S.I.2: S.I.≥ 150(EC150)最低刺激指数值
C1(μg/mL): S.I.1对应的受试物浓度
C2(μg/mL): S.I.2对应的受试物浓度
8、 结果解释
本试验结果能预测受试物的致敏性,但需应用合适的整合测试和评估方法(IATA)进一步评价受试物的致敏性,这些结果在有限的范围内外推到人类。
体外皮肤变态反应U937细胞系激活试验
起草说明
为加强化妆品的监督管理,进一步提高化妆品使用安全性,中国食品药品检定研究院组织开展了体外皮肤变态反应U937细胞系激活试验方法的起草工作。现就起草工作有关情况说明如下:
一、起草原则
试验要求和规定逐步与国际接轨,对于已经比较成熟的、被多数国家和组织认可的方法,原则上予以接受。对某些内容进行具体化和明确化,在尽量提高可操作性的同时,兼顾了技术发展的前瞻性。本方法制定过程中还注意了科学性和合理性、协调性和有效性,以及通俗性和规范性之间的关系。
二、起草过程
本研究由化妆品标准专家委员会于2018年立项,课题组在研究和分析国内外相关研究的基础上,对9种推荐物质进行了方法验证,并委托三家实验室进行实验室间验证,根据实验室验证过程和结果起草了试验方法文本。
三、与我国已有相关标准的关系
《化妆品安全技术规范》中用于检测皮肤变态反应的方法包括:皮肤变态反应试验、皮肤变态反应:局部淋巴结试验:DA(LLNA:DA)、皮肤变态反应:局部淋巴结试验:BrdU-ELISA(LLNA: BrdU-ELISA)、化妆品用化学原料体外皮肤变态反应:直接多肽反应试验(DPRA)。其中皮肤变态反应试验、LLNA:DA、LLNA: BrdU-ELISA均使用动物(豚鼠、小鼠)进行试验;DPRA方法针对皮肤变态反应AOP中的第一个关键分子事件进行检测。U937细胞系激活试验(U-SENSTM)针对皮肤变态反应AOP中的第三个关键分子事件进行检测。未来该方法可以应用在皮肤致敏评价的整合策略中,进一步提高替代试验方法的准确性和特异性。
本研究建立了皮肤变态反应的替代试验方法,以期与国际标准接轨,与《化妆品安全技术规范》中皮肤变态反应检测方法在技术上形成互补,建立我国动物实验替代方法体系,增补、修订和完善现有的标准与规范方法以及促进我国未来的化妆品安全评价体系提供技术支持。
四、与国际同类标准的关系
2016年该方法通过欧盟替代方法验证中心科学咨询委员会(ESAC)独立同行评议,2018年,经济合作和发展组织(OECD)正式发布U-SENS™方法的操作指南(OECD-442E),将其用于化学物质皮肤致敏性的安全评价。
本方法涵盖了OECD 2018年正式发布的“皮肤致敏反应有害结局通路中关于树突状细胞活化的关键事件的体外试验方法操作指南:U937细胞系激活试验”(OECD GUIDELINE FOR THE TESTING OF CHEMICALS In Vitro Skin Sensitisation assays addressing the Key Event on activation of dendritic cells on the Adverse Outcome Pathway for Skin Sensitisation ANNEXⅡ:U937 CELL LINE ACTIVATION TEST (U-SENS™))的要求,基本操作和OECD U-SENS™方法一致,具体变动内容包括:
(1)OECD方法中的阳性对照为“2,4,6-三硝基苯磺酸”属于危险化学品,因此,在本试验中增加阳性对照物质“松香酸”。
(2)在标准中对操作方法进行细化,如“铺板过程混匀防止细胞沉降”“轻轻涡旋清洗细胞”,以提高方法的可操作性。
各项技术内容的依据来源自OECD GUIDELINE FOR THE TESTING OF CHEMICALS In Vitro Skin Sensitisation assays addressing the Key Event on activation of dendritic cells on the Adverse Outcome Pathway for Skin SensitisationANNEXⅡ:U937 CELL LINE ACTIVATION TEST (U-SENS™)(OECD 《化学品测试指南》TG 442E,2018年),基本涵盖了其对采用对象、实验操作和结果分析的技术要求,并通过试验确认。
五、实验室验证结果
本方法实验室内验证结果和OECD参考值范围均一致;委托3家单位对方法进行实验室间验证。所有单位对8种化学物的定性预测结果均一致;4家实验室对8种OECD参考物质的定量检测结果(CV70、EC150)与OECD TG 442E附表中规定范围相比,准确率均为为100%。
化妆品禁用原料目录增补表
|
序号 |
中文名称 |
英文名称 |
|
1286 |
比马前列素 |
Bimatoprost(CAS No. 155206-00-1) |
|
1287 |
拉坦前列素 |
Latanoprost(CAS No. 130209-82-4) |
|
1288 |
他氟前列素 |
Tafluprost(CAS No.209860-87-7) |
|
1289 |
他氟乙酰胺 |
Tafluprost ethyl amide,(5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-Difluoro-4-phenoxy-1-buten-1-yl]-3,5-dihydroxycyclopentyl}-N-ethyl-5-heptenamide(CAS No. 1185851-52-8) |
|
1290 |
曲伏前列素 |
Travoprost(CAS No. 157283-68-6) |

来源:中检院


