前言
方法学验证的目的是证明所开发的方法能够满足检测的要求。检测的目的不同,对方法的要求也不同,验证的程度也就不同。进行方法学验证简单来说就是遵循官方指导原则(各国药典、CDE、ICH等),对方法的专属性、系统适用性、定量限、检测限、线性与范围、精密度、准确度、耐用性等进行验证,证明其符合相关要求。
由于各项指导原则只是宽泛的描述与规定,并没有具体操作和实施的细节,每个人对指导原则的理解不尽相同,因此对于方法学验证可以说是千人千面,各有各的特色。方法学验证并没有绝对的对与错之分,只要能客观合理的证明方法的可行性,就是合格的方法学验证。下面我将结合自己的方法学验证经验谈一谈对方法学验证的理解,探讨方法验证应该怎么做,为什么要这么做,欢迎各位同仁多多交流。
一、 需要验证哪些指标?
验证指标的确认与方法的性质相关,如对样品杂质限度检查进行验证只需对其专属性、检测限和耐用性进行验证即可满足限度检查需求,无需对准确度、精密度、检测限、定量限、线性与范围等与定量相关的指标进行验证。对于杂质的定量检测,则需进行专属性、准确度、精密度、检测限、定量限、线性与范围、耐用性等一系列的验证方能证明其用于杂质定量测定的可靠性。不论是哪种检测需求,专属性和耐用性验证是必不可少的,这也是每一个分析方法必须满足的基本要求。具体的验证指标设置可参照下表1。
▲ 表1-检验项目和验证指标
|
指标/项目 |
鉴别 |
杂质测定 |
含量测定—特性参数—含量或效价测定 |
|
定量 |
限度 |
|
专属性b |
+ |
+ |
+ |
+ |
|
准确度 |
- |
+ |
- |
+ |
|
精密度 |
|
|
|
|
|
重复性 |
- |
+ |
- |
+ |
|
中间精密度 |
- |
+a |
- |
+a |
|
检测限 |
- |
-c |
|
- |
|
定量限 |
- |
+ |
- |
- |
|
线性 |
- |
+ |
- |
+ |
|
范围 |
- |
+ |
- |
+ |
|
耐用性 |
+ |
+ |
+ |
+ |
a:已有重现性验证,不需要验证中间精密度
b:如一种方法不够专属,可用其他分析方法予以补充
c:视具体情况予以验证
二、 方法验证内容
2.1 专属性
专属性指在杂质、降解产物、辅料等其他组分可能存在的情况下,采用的分析方法能正确测定出被测物的能力。对于专属性不强方法,需采用不同原理的方法予以补充。
在方法学验证过程中专属性考察的方法包括定位试验和降解试验。
2.1.1 定位试验
在杂质对照品和辅料可以获得的情况下,对于含量测定,可向供试品中加入杂质或者辅料,与未加杂质或者辅料的供试品测定结果比较,以此考察测定结果是否受干扰;对于杂质检查,可向供试品中加入一定量的杂质,考查杂质之间分离度是否符合要求。
在做定位试验时需要将各杂质或辅料单独进样,再将杂质或辅料与API混合后进样,考察杂质及辅料是否干扰目标组分的测定。通常情况下,混合溶液中各杂质的保留时间和峰面积与单独定位时基本一致,但有时也会发生保留时间漂移或峰面积减小且同时产生新的杂质峰的情况,对于保留时间漂移,最可能原因是受到了供试品溶液基质的影响,由于实际情况下杂质与供试品总是同时存在的,因此需以混合溶液中杂质的保留时间为准,而不以单独定位时的保留时间为准。对于峰面积减小且产生新杂质的情况我们需要确定是哪些组分发生反应,做定位时需现配现进或将发生反应的组分分成两个混合溶液分别进行考察,避免反应的发生。
2.1.2 降解试验
降解试验的目的是通过强制降解来研究潜在的降解杂质和降解途径对含量和杂质测定的影响,以进一步考察方法的专属性。降解程度建议主峰归一化纯度降低5%~10%,如果通过很剧烈的条件供试品仍未发生明显降解那么说明供试品很稳定,在日常的运输储存和使用过程中不会发生难以预料的降解,因此无需进一步降解。降解试验完成后应统计新增杂质,同时对新增杂质进行LC-MS研究,结合API结构特征和降解条件推测其结构。
专属性要求:
1)在专属性试验中各杂质及辅料不干扰目标组分的测定,待考察组分与相邻峰分离度大于1.5。
2)各降解杂质不干扰目标组分的测定,降解物料守恒90%~110%之间(仅有关物质)。
3)定位及降解试验中目标组分的色谱峰纯度符合要求。
2.2 系统适用性
与仪器核查中的系统适用性不同,方法学验证中的系统适用性是将仪器设备、试验操作和被分析样品作为一个整体来考察。凡是样品检测中需要用到的溶液(如有关物质测定中的自身对照溶液、供试品溶液、混合溶液,含量测定的对照品溶液、供试品溶液等)均需要进行系统适用性考察。
系统适用性考察时将待考察溶液连续6针进样,计算保留时间、峰面积或杂质含量RSD,同时考察待测组分的理论塔板数和拖尾因子及与相邻峰的分离度是否符合要求。通常保留时间RSD≤1%,峰面积RSD≤2%,杂质含量RSD可参照药典四部9101方法学验证指导原则精密度项下要求(见下表2)来设置。待测组分与相邻杂质分离度不小于1.5,理论塔板数可依据样品及色谱柱的特点来设置,一般C18色谱柱理论塔板数会高些,SEC色谱柱塔板数会稍差,此外小分子化合物的理论塔板数高些,生物大分子理论塔板数低些,这个只要在规定的理论塔板数下能够满足专属性、灵敏度或准确性等验证指标的要求即可,没有统一的标准。拖尾因子一样也没有固定的要求,药典中指出对于采用峰高计算的方法应尽量将拖尾因子控制在0.95~1.05之间。在实际的验证中通常都是采用峰面积计算,因此对拖尾因子的要求可适当放宽,尤其是有关物质检测,在其他指标符合要求的前提下拖尾因子可进一步放宽。
2.3 检测限
检测限是指供试品中被测组分能被检测出的最低量。检测限的测定方法包括直观法、信噪比法和基于响应值标准偏差和标准曲线斜率法,最常用的是信噪比法,要求检测限信噪比≥3。
2.4 定量限
定量限是指供试品中待测组分能被定量测定的最低量。定量限的测定方法包括直观法、信噪比法和基于响应值标准偏差和标准曲线斜率法,最常用的是信噪比法,要求定量限信噪比≥10。
在验证方法的定量限时还需验证定量限精密度及回收率。
定量限精密度是平行配制6份定量限溶液,计算6份溶液待测组分峰面积RSD,RSD限度要求参见精密度项下表2。
定量限回收率是向供试品中添加定量限水平的杂质对照品(平行配制6份),外标法计算其回收率并计算回收率RSD,回收率限度要求参见准确度项下表3。
2.5 线性
线性关系是定量分析的基础和必要条件,具体做法是采用对照品配制一系列浓度的溶液,有关物质浓度设置范围通常为定量限~限度200%,采用归一化计算时主成分需做到100%供试品浓度;含量测定的线性范围至少包含80%~120%,也可以根据需要放宽范围,但要保证80%、100%和120%在线性考察点上。线性考察至少要设置5个浓度,然后以浓度为横坐标,峰面积为纵坐标作线性回归,记录线性方程、相关系数、线性图、残差平方和,除此之外还应考察Y轴截距和响应因子。通常相关系数应大于0.999,截距应不大于100%相应2%(含量)或25%(杂质含量),响应因子RSD不大于10%。
校正因子:校正因子表示主成分与杂质单位浓度相应的比值,通常是采用主成分与杂质线性斜率的比值计算,计算校正因子允许的误差通常为10%,需由不同试验人员在不同的仪器上进行测定,以增加其准确性和耐用性。通常校正因子在0.9~1.1可以采用不加校正因子的方法计算杂质含量;当校正因子超出这个范围但在0.2~5.0时可以采用加校正因子的方法计算杂质含量;当校正因子<0.2或>5.0时需采用外标法计算杂质含量。校正因子小于1时保留两位小数,大于1时保留一位小数,也就是保留两位有效数字。举个例子:如主成分线性斜率为56000,杂质1线性斜率为59000,杂质2线性斜率为52000,那么杂质1的校正因子是0.95,杂质2的校正因子是1.1。
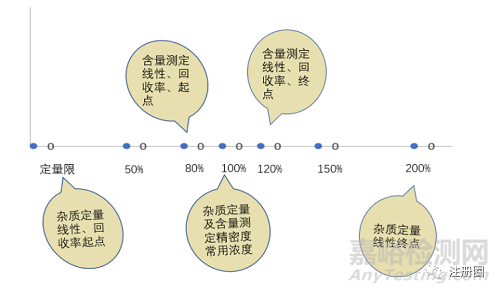
2.6 范围
范围是指所开发方法能达到符合要求的精密度、准确度和线性时的高低限浓度或量的区间。不同的验证指标范围设置不同,通常线性≥准确度≥精密度。推荐的范围设置见下图1。
▲ 图1-线性、精密度及回收率通常设置的范围
2.7 精密度
精密度指同一份均匀供试品在规定测定条件下经多次取样测定所得结果之间的接近程度,一般用偏差、标准偏差或相对标准偏差表示。精密度可以分为三个层次,重复性是在相同条件下,由同一个分析人员测定所得结果之间的精密度;中间精密度是在同一实验室内发生分析人员、检测时间、仪器设备等随机变动情况下所获得测定结果之间的精密度;重现性是不同实验室测定结果之间的精密度。
精密度考察有两种方式,一是在规定范围内,取至少6份同一浓度(相当于测定浓度100%水平)供试品的测定结果进行评价;二是设计不少于3种不同浓度(通常为测定浓度80%、100%、120%水平),每种浓度配制不少于3份供试品溶液进行测定和评价。目前第一种做法是普遍的做法。精密度结果的评价标准参照下表2设置。
▲ 表2-样品中待测定成分的含量与精密度可接受范围关系
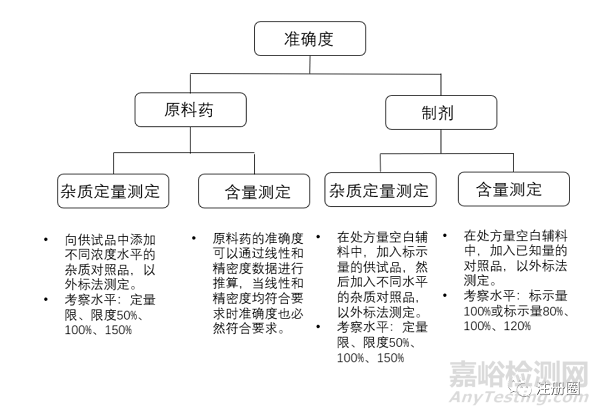
2.8 准确度:
准确度一般用回收率来表示,表示方法测定值与真实值的接近程度,准确度的测定方法参见下图2。
▲ 图2-准确度测定方法
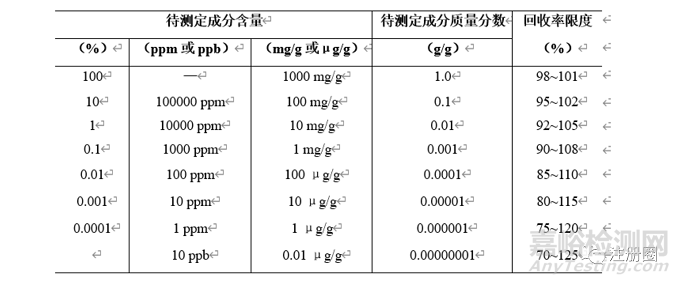
准确度的限度设置可以参考下表3,当基质较复杂或待分析组分含量较低时也可适当放宽限度。
▲ 表3-样品中待测定成分的含量和回收率限度
2.9 耐用性
耐用性考察包含两部分内容,一是溶液稳定性,二是色谱条件波动对检测结果的影响。
2.9.1 溶液稳定性
溶液稳定性影响应用此方法所获得数据的准确性、可靠性、有效性。凡是样品检测中需要用到的溶液(如有关物质测定中的自身对照溶液、供试品溶液、混合溶液,含量测定的对照品溶液、供试品溶液等)均需要进行稳定性考察,以确保样品检测的可靠性。在进行溶液稳定性考察时首先应考察25℃的稳定性,如稳定性较差可考察更低温度下的稳定性,此外如果25℃稳定性极差应注意样品临用新配或者考虑更换配制溶剂。考察的时间可根据需要或早期稳定性的结果来设置。溶液稳定性的评价可以采用偏差、平均偏差或相对标准偏差。对于含量测定的供试品溶液、对照品溶液或有关物质的自身对照溶液可以比较峰面积的变化,对于有关物质的供试品溶液则应考察单个杂质含量、总杂质含量、杂质个数等多个指标。
2.9.2 色谱条件耐用性
指测定结果不受测定条件微小变动影响的承受程度,考察耐用性可以为所建立的方法用于常规检验提供依据。通常方法开发过程中就应考虑其耐用性,如果测试条件要求苛刻,应在方法中注明可以接受范围。
色谱条件耐用性考察常见的波动范围如下表4:
▲ 表4-耐用性色谱条件变化示意表
耐用性中色谱条件的变化范围可以根据方法实际的耐用性作调整。在耐用性考察中除了考察含量、杂质含量等,重要的一点是要考察各耐用性条件下的专属性如何,可以通过考察各耐用性条件下混合溶液中待测组分与相邻峰的分离度来评价。
三、 小结
依据QbD理念,药品不是检验出来的是设计出来的,与药品一样,分析方法不是验证出来的,是设计出来的。在方法开发的过程中就应充分考虑后续方法验证的可行性,建立稳健性较好的方法,为方法验证提供一个较好的输入。
其次,方法学验证与所建立的质量标准是紧密相关的,当质量标准发生变更如杂质限度修订、残留溶剂种类、生产工艺或制剂处方调整等都可能涉及到方法学的部分或重新验证。在开展方法学验证的过程中要理清到底为什么做方法学验证,做哪些试验可以证明方法的可靠性,怎样合理设置各项指标的控制限度,不要为了验证而验证,也不要做得千篇一律,要根据当前研究阶段对样品质量控制的需求进行验证,既不要过于粗犷也不要过度验证。以上是我对于方法学验证的一点个人看法,欢迎各位药学同仁交流探讨。
参考文献:
[1] 中国药典 2020 版四部通则9101“分析方法验证指导原则”
[2] ICH Q2(R1) Validation of Analytical Procedures Text and Methodology
[3] 化学药物质量控制分析方法验证技术指导原则