摘要
目的:分析2011—2021年期间中国获批新药上市的绝对滞后与相对滞后的状况及原因,并初步探究影响新药上市滞后的潜在因素。
方法:收集2011—2021年期间中国、美国、欧盟以及日本药监部门批准上市的新药信息,并确定中国批准上市的新药在全球首次获批上市的时间,以计算中国新药上市的绝对滞后和相对滞后;采用多元回归分析方法探究药物滞后相关的影响因素。
结果:2011—2021年期间中国共批准新药280个,与美国、欧盟和日本的绝对滞后分别为180,82和154个。总体相对滞后中位时长为1359d,其中2011—2014年滞后中位时长逐渐增加,2014年达到顶峰(3438d);2015—2018年滞后中位时长下降明显;2019—2021年滞后中位时长呈逐年下降态势,2020和2021年处于总体滞后中位时长以下,分别为1134和500d。感觉器官治疗领域[非标准化系数(B)=-1153.840,P<0.05]的新药与较短的滞后中位时长相关,具有孤儿药认定资格(B=441.147,P<0.01)、分类为生物制品(B=502.205,P<0.01)的新药与较长的滞后中位时长相关。
结论:2011—2021年中国新药上市滞后问题得到明显的改善,但各治疗领域的新药仍普遍存在滞后现象。在新政策背景下,进一步提升新药审评审批的质量和效率、加强政府审批机构与申报企业的沟通交流、对接国际通行规则、鼓励企业开展国际多中心临床试验等是减少新药上市滞后的关键。
关键词:上市滞后;新药审评审批;滞后因素
正文:
新药上市是一个药物从研发到商业化的重要里程碑,意味着患者能够获得最新的治疗方案,对改善病种的发病率和死亡率都有着重要的意义。然而,自20世纪60年代“反应停”事件发生以来,各国政府纷纷加强了对新药上市的监管,制药企业需要进行临床试验证明药物的安全性和有效性,并经过药品监管部门的审评审批才能将药品上市销售。与新药相关的不良反应事件发生往往会让监管部门对新药的审评审批更加严苛谨慎,加上各国药品监管系统的差异以及药企研发策略的不同,导致了同一个新药在不同国家上市的批准时间具有先后顺序,这一现象被称为“药物滞后(drug lag)”[1]。
中国的药物滞后问题已存在多年,过去20多年中国新药获批上市的数量远远落后于欧盟、美国等发达国家或地区[2],国家药品监督管理局药品审评中心(Center for Drug Evaluation,CDE)的注册申请积压影响了具有高临床治疗价值新药的可及性。虽然中国药品监管部门自2009年起出台了一系列如《药品注册特殊审批管理规定》、《关于药品医疗器械审评审批制度的意见》、《化学药品注册分类改革工作方案》、《关于解决药品注册积压实行优先审批的意见》等政策文件,旨在加快新药审批审评、解决注册审批积压问题,并取得了不错的成效,但是中国新药上市滞后问题依旧存在[3-6],2016—2020年中国批准的进口抗肿瘤新药上市时间相对于美国滞后中位时长为2.7年[7]。因此,本研究以2011—2021年在中国获批上市的新药为研究对象,以药物国际诞生日(international birth date,IBD)作为时间标准分析中国新药批准上市的滞后问题并对其影响因素进行初步探究,探寻新药上市滞后的关键原因,为解决中国药物滞后问题提供参考。
1、数据来源与方法
1.1 IBD
IBD是指药品在世界上任何国家首次上市许可的日期[8]。
1.2 数据收集及纳入标准
由于世界各国对新药的定义各不相同,为保证新药的纳入标准一致,本研究以国际监管科学创新研究中心的研究作为参考[9],将新药定义为含有新分子实体(new molecular entities,NMEs)的药物。其中根据ICH指导原则Q1A(R2)部分,新分子实体是指在有关国家或地区的药品管理机构注册过的任何药品中从未出现过的一种活性药物成分。
国内药品信息数据主要来源于第三方数据库“Insight”数据库[10]、“药融云”数据库[11]、国家药品监督管理局(National Medical Products Administration,NMPA)和CDE官网公开信息、CDE药品年度审评报告[12];美国药品信息数据主要来源于Drug@FDA数据库[13]和美国FDA官网公开信息;欧盟药品信息数据主要来源于欧洲公共评估报告(European Public Assessment Report,EPAR)[14]、成员国药品信息登记目录(National Registers,NR)[15]及欧洲EMA官网公开信息;日本药品信息数据主要来源于药品和医疗器械监督管理局(Pharmaceuticals and Medical Devices Agency,PMDA)官网“已批准的产品列表”[16]。
从以上数据库(中国的新药数据主要来自“Insight”数据库)收集2011—2021年批准上市的化学新药和生物新药(治疗用生物制品)的相关信息,包括药品名称、获批适应证、批准时间、企业名称、药品类型等。因为中国分别在2016和2019年对化学药品和生物制品的注册分类进行修订,所以中国化学新药数据纳入标准分为2016年以前旧注册分类为1.1类、1.2类、1.5类、3类和2016年以后新注册分类为1类、5.1类的新药;生物新药数据纳入标准分为2020年以前注册分类为1类、2类、3类、4类、6类、7类和2020年以后注册分类为1类、3.1类、3.2类的新药,同时需要对上述分类的化学药和生物药进行筛选,通过“药融云”数据库、NMPA官网公开信息进一步排除当中不符合本研究新药定义的药物。IBD通过“药融云”数据库、全球各国的药监政府官网信息、药企官网信息进行确定,对于一些争议性新药的IBD将进一步通过学术文献、新闻、行业报告等资料加以确认,若仅提及批准年份则批准日期统一按该年1月1日处理。本研究统计范围不包括中药、预防用生物制品(疫苗)、诊断试剂以及药械组合产品。
1.3 药物滞后分析
药物滞后分为绝对滞后和相对滞后,前者可用批准药物的实际数量进行衡量,即一段时间跨度内不同国家/地区批准上市的药物数量差异;后者衡量的标准是时间,即某一药物在不同国家/地区之间批准上市的时间差[17]。绝对滞后以中国NMPA、美国FDA、欧洲EMA和日本PMDA这4个国家/地区的监管机构批准的新药数量进行计算,相对滞后以新药首次全球批准时间作为标准,计算中国每个上市新药获批的滞后时间,以中位时长进行分析。本研究将新药获批上市的类型分为“中国首批”、“国外首批”、“仅境内获批”以及“境内外获批”。“中国首批”是指新药全球首次上市获批发生在中国,IBD为在中国获批上市的日期;“国外首批”是指新药全球首次上市获批发生在国外,IBD为在国外获批上市的日期;“仅境内获批”是指新药只在本国或地区境内获批上市,境外尚未有国家或地区批准许可上市;“境内外获批”是指新药除在本土获批以外,在其他国家/地区(不包括中国香港地区、中国澳门地区和中国台湾地区)同样获批上市。厂家类型统计分为“国外企业”和“国内企业”,其中“国外企业”是指由外商独资经营的制药企业;“国内企业”是指内资制药企业,统计包括中外合资企业以及从外国企业获得药品许可的内资企业。“孤儿药认定资格”以美国FDA认定的孤儿药为准;若药物未获得美国FDA孤儿药认定资格,则以中国卫生健康委员会发布的《第一批罕见病目录》(以下简称《罕见病目录》)作为依据[18],根据药物上市获批的适应证进行孤儿药界定。
1.4 统计分析
由于数据不呈正态分布,因此使用Wilcoxon符号秩和检验来检验统计结果的显著差异。多元回归分析作为统计学上分析数据的方法,目的在于了解多个自变量与因变量之间是否具有线性关系,预测药物滞后可能受多个因素影响,因此使用多元回归分析探索与药物滞后相关的因素。以上均使用SPSS Statistics 26.0统计软件对数据进行分析。
2、结果
如表1和表2所示,2011—2021年间,中国共批准280个新药上市,其中化学药203个(72.5%)、生物药77个(27.5%)。
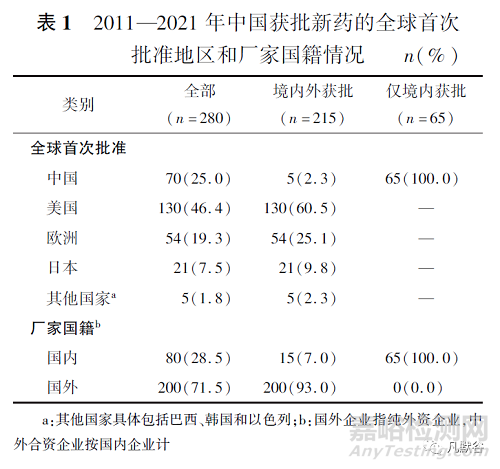
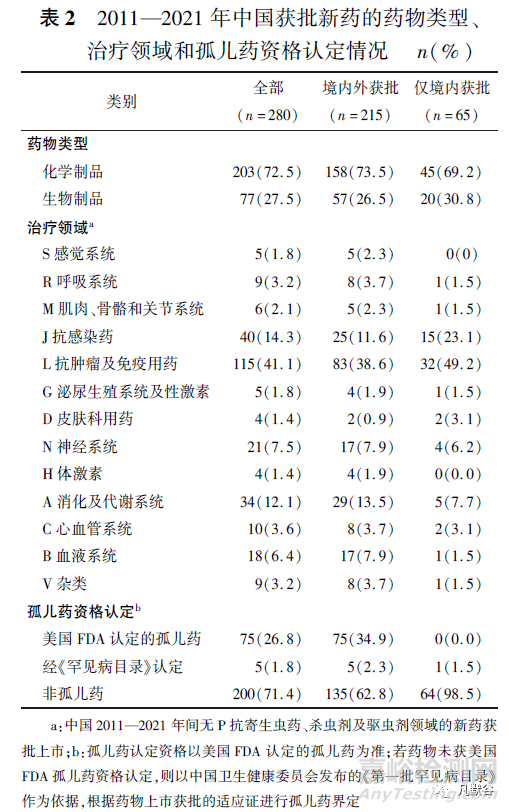
在280个新药中,有70个新药是由中国全球首次批准,占所有获批新药的25.0%,其余分别为美国全球首批130个(46.4%)、欧洲地区54个(19.3%)、日本21个(7.5%)、其他国家5个(1.8%)。在中国全球首次批准上市的70个新药中,已在其他国家批准上市的新药仅5个(艾拉莫德、罗沙司他、西达本胺、康柏西普、艾博韦泰),其余所有新药(65个)只在中国境内上市,并且均来自国内企业申报。中国国内企业和国外企业的新药获批数量存在较大差异,国外企业获批上市的新药个数为200个,占中国所有获批新药数量的71.5%;国内企业获批上市的新药个数为80个,占28.5%,其中15个取得国外监管机构的上市许可。
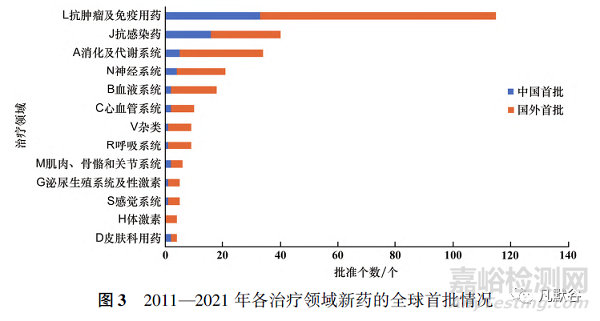
另外,国内企业获批上市的新药数量多于中国全球首次批准数量,说明了部分国产新药全球首次获批上市地点是在国外而非中国。按世界卫生组织药物解剖学、治疗学及化学(anatomical therapeutic chemical,ATC)分类代码第一级治疗领域划分[19],L抗肿瘤及免疫用药(115个,41.1%)、J抗感染(40个,14.3%)、A消化及代谢系统(34个,12.1%)是中国新药批准活跃的三大治疗领域,对于仅本土获批的新药而言,L抗肿瘤及免疫用药领域的新药占近一半(33个,48.5%)。相对地,过去11年中国较少新药获批上市的治疗领域是H体激素(4个,1.4%)和D皮肤科用药(4个,1.4%)。按孤儿药资格认定进行统计,共80个孤儿药获批上市,约占所有获批新药数量的1/4。
2.1 中国新药上市的绝对滞后
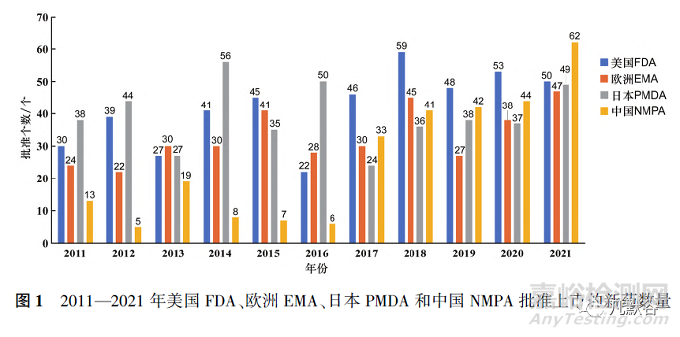
图1显示了美国、欧盟、日本和中国这4个国家/地区于2011—2021年间上市的新药数量,各药品监管机构批准新药数量由多至少分别是美国FDA(460个)、日本PMDA(434个)、欧洲EMA(362个)、中国NMPA(280个)。
11年间中国NMPA相对于美国FDA、日本PMDA、欧洲EMA批准新药的绝对滞后分别为180,154和82个,2017年起中国NMPA与欧洲EMA和日本PMDA每年的绝对滞后基本为0,但与美国FDA仍存在滞后(见表3)。
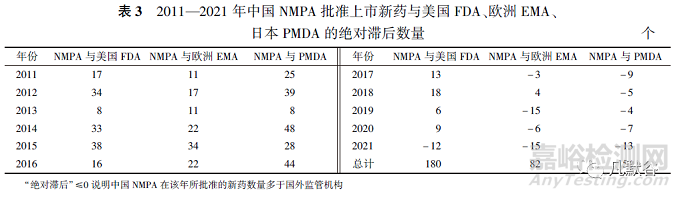
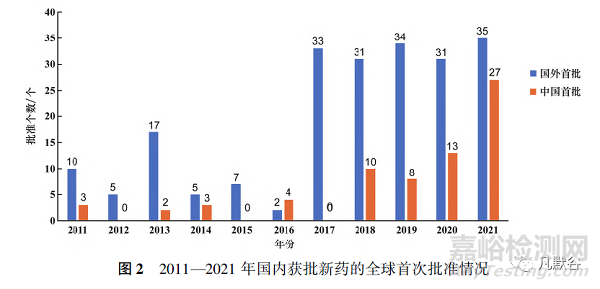
中国每年批准新药数量在2011—2016年期间远远落后于其他3个国家/地区,2014—2016年连续3年仅有个位数的获批新药数量,但2017年以后中国批准新药数量井喷式爆发并且逐年上升,2021年批准新药数量达到了62个,为历年最高。此外,2017年后每年“国外首批”的新药数量总体维持在30个左右,而“中国首批”的新药数量却在不断增加,2021年“中国首批”与“国外首批”的新药数量仅相差8个(见图2)。
在13种ATC治疗领域一级分类中,同样是以“国外首批”的新药上市为主,获批新药主要集中在L抗肿瘤及免疫用药领域,获批新药数量远超第2位和第3位的J抗感染药和A消化及代谢系统,在H体激素领域中没有诞生“中国首批”的新药(见图3)。
2.2 中国新药上市的相对滞后
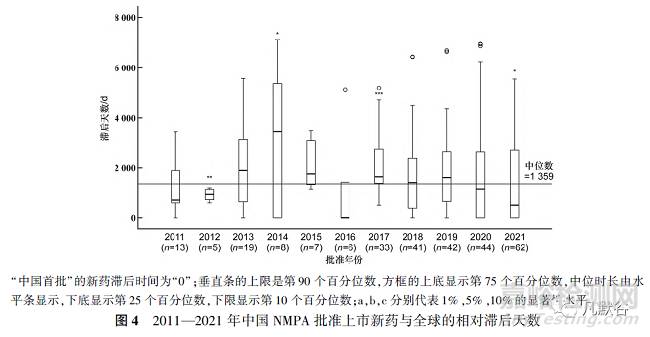
通过计算新药获批上市日期与IBD的时间差得到2011—2021年中国NMPA批准上市新药相对于IBD的滞后天数,并使用Wilcoxon符号秩和检验比较每年新药获批上市的相对滞后天数与总体滞后中位时长的差异,结果见图4。新药上市总体滞后中位时长为1359d,自2011年起中国新药批准上市滞后中位时长逐渐变大,于2014年达到顶峰(3438d),随后几年逐渐下降,2020和2021年滞后中位时长明显低于总体滞后中位时长,分别为1134和500d。
由于2014年滞后中位时长达到峰值,故将2011—2014年、2015—2018年、2019—2021年分为3组以观察不同治疗领域间的上市滞后变化,0≤滞后时间中位数(年)<2为“+”,2≤滞后中位时长(年)<4为“++”,4≤滞后中位时长(年)<6为“+++”,滞后中位时长(年)≥6为“++++”,见表4。
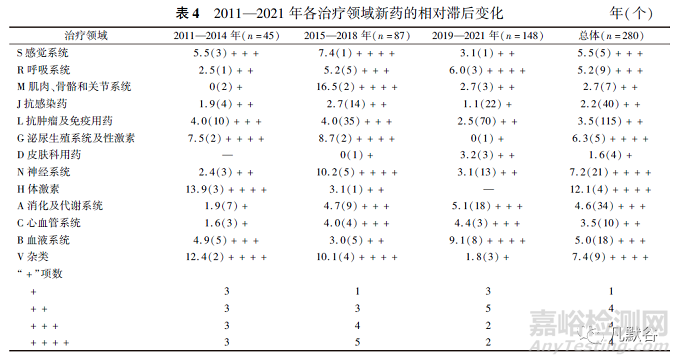
总体来看,所有新药中,感觉系统、抗肿瘤及免疫用药、抗感染药、泌尿生殖系统及性激素、神经系统等治疗领域的滞后中位时长在近11年来均有所改善或保持良好,在新药批准数量相对较多的治疗领域中(总批准数量≥20个),仅消化及代谢系统领域的滞后中位时长在增加,3组滞后中位时长分别为1.9,4.7和5.1年。对于2019—2021年批准的药物,滞后中位时长≥4年的治疗领域有4项,分别是消化及代谢系统(5.1年)、心血管系统(4.4年)、呼吸系统(6.0年)和血液系统(9.1年),相比总体或者2011—2014年和2015—2018年所批准的药物,滞后中位时长在4年以上的治疗领域项数已明显减少。
2.3 中国新药上市滞后影响因素的初步探究
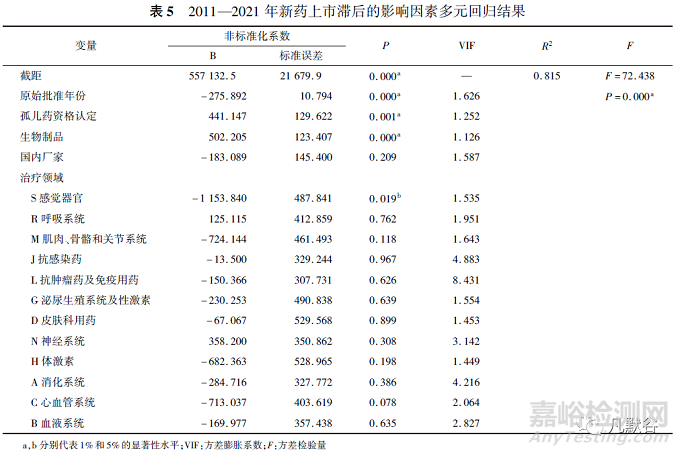
国外有研究指出,药物滞后与几个因素有关,包括企业国籍、分子类型、治疗领域、孤儿药地位、审评过程和临床研发的策略[如纳入全球临床试验(MRCT)][20-21]。为识别中国新药批准上市滞后的影响因素,以“相对滞后时间”作为因变量,“厂家国籍”、“药物分子类型”、“治疗领域”、“孤儿药资格认定”、“原始批准日期(按年份计)”作为自变量并创建分类变量进行多元回归分析,其中“厂家国籍”、“药物分子类型”以及“孤儿药资格认定”为二值变量,赋值为0,1;“治疗领域”为多分类变量,以“V杂类”作为参照组共设置12个哑变量。回归结果见表5。
整体回归水平上呈显著性(F=72.438,P<0.01)。结果表明,新药上市滞后时间与原始批准年份(B=-275.892,P<0.01)、感觉系统治疗领域(B=-1153.840,P<0.05)呈负相关;与具有孤儿药资格认定(B=441.147,P<0.01)、药物分类为生物制品(B=502.205,P<0.01)呈正相关。尽管不同治疗领域之间的新药上市滞后时间存在差异,但除了感觉器官领域外,回归结果未能说明不同治疗领域分组和滞后中位时长之间存在相关性;新药上市滞后时间与“国内企业”呈负相关,这是由于国内企业获批的新药相对滞后时间绝大部分为0,理论上应能降低药物滞后时间,但回归结果无统计学意义,因此本研究数据尚不能证实厂家国籍是影响新药上市滞后的重要变量。
3、讨论
本研究比较了2011—2021年间美国、欧盟、日本、中国4个国家或地区批准的新药数量以及在中国获批上市的280个新药的获批时间与其在全球首次获批的时间差异。结果表明,同一时间跨度内,中国NMPA批准上市的新药总数量远远落后于美国FDA、欧洲EMA和日本PMDA,获批上市时间相对于IBD而言,总体滞后中位时长为1359d。
3.1 药品审评审批制度改革初显成效,新药上市滞后问题得到改善
过去冗长的药品审评审批和临床试验审批程序是造成药物滞后的原因之一[22],但近年来得益于药品审批审评制度改革工作的持续推进,中国新药上市的滞后问题正逐步好转。从绝对滞后的角度来看,2017—2021年间中国NMPA与欧洲EMA、日本PMDA的绝对滞后基本为0,2017年无疑是中国新药上市绝对滞后减少的转折点。一方面,原国家食品药品监督管理总局(CFDA)在2017年正式加入ICH,这意味着中国在药品注册技术方面的要求与国际规则逐步接轨。对企业而言,按照国际通行规则进行新药研发更容易获得其他国家的批准上市,不仅有利于进口新药进入中国市场,同时也有利于国产新药面向国际;对NMPA而言,接受并参与国际规则的制定和实施能减少中国与其他国家之间在药品注册技术、申报资料等要求上所存在的审评差异,从而提升新药的审评审批效率;另一方面,原CFDA于2017年出台的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》和《国家食品药品监督管理总局关于调整进口药品注册管理有关事项的决定》中进一步放开了国际多中心临床试验数据在新药注册申报时的使用限制,鼓励企业开展全球同步的临床试验,并优化了进口新药的注册申报程序,此举减少了非必要的重复申报和临床试验,从而加快了新药在中国的上市速度。从相对滞后的角度来看,新药获批上市的滞后中位时长在2011—2014年间伴随着CDE审评任务的大量积压达到峰值,自2015年以来,原CFDA出台《总局关于发布化学药品注册分类改革工作方案的公告》、《总局关于发布化学药品新注册分类申报资料要求(试行)的通告》、《关于解决药品注册申请积压实行优先审评审批的意见》等一系列政策文件来解决审评积压问题,具体措施包括对化学药品的注册分类、注册申报资料、优先审评审批等重新作出要求,新修订的申报资料要求更加趋向于国际通用的标准注册技术文件格式(CTD),进一步规范了注册申请人按照标准的新药研发理念进行上市前的研究[23],也使得2015年之后的滞后中位时长有了明显下降。此外,2019年新修订的《药品管理法》中新增的附条件审批制度、临床试验默示许可制度、关联审评审批制度等在加快新药上市速度方面都起到了重要作用,自2019年《药品管理法》以及2020年《药品注册管理办法》实施以来,2021年的滞后中位时长下降到500d,为过去11年中最低。
3.2 新药以欧洲、美国全球首批居多,国产新药受国际认可程度较低
中国批准上市的新药以境外已上市的新药为主,其中大部分新药由美国FDA或欧洲EMA率先批准上市,随后引进至中国。由中国药品监管部门批准首次全球上市的新药有70个,占所有批准上市新药的25.0%,并且几乎全部来自本土企业的申报。同样的,近几年来日本也存在相似情况,Tanaka等[24]在关于日本新药上市滞后问题的研究中发现2008—2019年在日本批准上市的400个新药中有80个(20%)是由日本PMDA批准首次全球上市,其余大部分新药都来自欧洲或美国。此外,在由中国批准首次全球上市的新药当中只有5款在国外获得批准上市,分别是2011年上市的艾拉莫德片、2013年上市的康柏西普眼用注射液、2014年上市的西达本胺片、2018年上市的罗沙司他胶囊和艾博韦泰注射液,其余65款国产新药仅在本土获批;相比之下,日本在这方面的新药多达35款。可见,全球首次上市的新药以诞生在欧洲、美国的居多,虽然由中国首批全球上市的新药具有一定比例,但其中能走出国门获得其他国家批准上市的新药却少之又少。在全球一体化的大背景下,国产新药“出海”是本土医药企业谋求国际化发展的一个重要方向,但近年来却屡屡受阻。如2022年5月国内某药企的治疗胰腺和非胰腺神经内分泌瘤新药因临床研究数据不足被美国FDA拒绝批准上市,美国FDA在回复函中表明,需要纳入更多代表美国患者人群的国际多中心临床试验来支持美国获批[25];同一时间国内另外一药企的肿瘤免疫新药以需要进行质控流程变更为理由被美国FDA拒绝[26]。由于各国药品监管法律法规体系不一且复杂,国内企业想要立足国际化市场,必须要熟悉各国家的法律法规、紧跟当地最新的评审要求并依照国际标准行事,如布局全球多区域多中心临床试验、践行ICH E17指导原则、与监管部门开展及时、充分的沟通,这样在一定程度上可以减少各国药监体系差异带来的不确定性,为新药“出海”铺平道路,提高国产新药的国际认可度。
3.3 抗肿瘤新药研发火热,滞后问题有待进一步改善
本研究中,无论是在中国首批还是国外首批,抗肿瘤及免疫用药(115个)是中国新药上市最活跃治疗领域,相比于其他热门的治疗领域如消化及代谢系统(4.6年)、神经系统(7.2年),抗肿瘤及免疫用药的新药总体滞后时间虽然较短(3.5年),但仍长于抗感染药领域(2.2年)。近年来,抗肿瘤及免疫用药类新药拥有比其他治疗领域药物更多的研发资源和更高的优先地位。恶性肿瘤是慢性病患者的主要死因之一[27],21世纪以来逐渐成为世界新药研发的焦点[28],国内抗肿瘤类新药更是备受国际医药投资人的关注,2013—2018年间中国新药热点投资在抗肿瘤及免疫用药治疗领域占据了半壁江山[29]。自2016年2月实行优先审评审批制度以来,至2017年底为止,抗肿瘤及免疫用药领域类药物在所有纳入优先审评审批的药物类型中占比最大[30],原CFDA于2017年12月发布的《关于鼓励药品创新实行优先审评审批的意见》中也正式把具有明显临床优势且用于恶性肿瘤的药物纳入优先审评审批的范围[31]。巨大的市场空间、政策扶持力度以及资本的推动让2019—2021年抗肿瘤及免疫用药类新药的滞后中位时长减少至2.5年,但也导致了目前抗肿瘤及免疫用药类新药存在靶点高度集中、重复过度申报、新药同质化等问题[32]。因此,企业应该贯彻《以临床价值为导向的抗肿瘤药物临床研发指导原则》[33],坚持以“临床价值为导向”进行抗肿瘤新药的研发,投入到真正意义上的创新而非低效同质的“伪创新”,减少不必要的资源浪费,从而提升临床试验的质量和申报审评的效率,以进一步减少抗肿瘤新药的上市滞后时间。此外,抗肿瘤新药研发难度大,对方法学和临床观点的审评可能因监管机构而异,针对少数目标患者群体的招募困难、临床研究设计困难以及复杂的医学伦理等问题也可能是这类新药的上市滞后时长未能进一步下降的原因[34-35]。
3.4 孤儿药滞后问题较严重,现行激励政策作用不明显
多元回归分析的结果表明,中国新药上市滞后时间的增加与新药具备“孤儿药资格认定”有显著相关性(P≤0.01)。虽然过去10年中国采取了如优先审评、减轻增值税、减免临床试验等一系列激励政策以加快孤儿药上市[36-38],但是回归结果表明具备孤儿药资格的新药上市滞后时长仍会增加,现有激励措施对改善孤儿药上市滞后的作用尚不明显。罕见病相关政策以及企业研发起步较晚可能是主要原因之一。一方面,企业的研发决策可能是影响孤儿药上市滞后的一个因素。对于孤儿药而言,罕见病的患病率越低,市场潜力就越低,加上高昂的研发成本,导致企业对孤儿药研发的积极性不高。在Nakayama等[20]关于日本与美国的孤儿药滞后研究结果表明,日本与美国之间的孤儿抗癌药上市滞后中位时长为727d,其中上市滞后的主要影响因素是企业提交审评的时间滞后而非审评过程的时间滞后,企业提交审评的时间与企业与外部合作以及研发策略相关。另一方面,虽然中国早在1999年便提出了将孤儿药纳入特殊审批,但直到2018年国家卫生健康委员会才制定了罕见病种的纳入标准和目录,针对罕见病用药的法律法规、管理规定及实施办法仍有待建立;相比之下,美国、欧盟和日本分别于1983,1999,1993年就已建立了相对完善的孤儿药认定标准、审批监管程序以及法律制度[39-40]。此外,中国孤儿药主要依赖进口,本研究中具有国内外孤儿药资格的品种占28.6%,几乎全部为进口药,国产孤儿药数量为1,加强国内企业的孤儿药研发能力同样是改善孤儿药上市滞后的关键。
4、结论
本研究表明,中国药品审评审批监管改革效果显著,过去2011—2021年中国药物滞后问题得到明显改善,具体表现为每年批准新药的数量上升、相对药物滞后中位时长自改革后下降,但是各个治疗领域类型的新药上市仍存在药物滞后现象,包括孤儿药和生物制品。此外,提升新药审评审批的质量和效率、加强政府审批机构与申报企业的沟通交流、对接国际通行规则、鼓励企业开展国际多中心临床试验是减少新药批准上市滞后的重要手段,对于新药“走进来”或是“走出去”都可能有积极影响。
参考文献
《中国新药杂志》 2023年第32卷第11期