第1部分:器械描述和规范,包括器械的变体版本和附件
预期用途
技术文件所包含的器械
分类
器械描述和规范说明
器械描述应能够让人理解器械的设计、构成和表现以及其他特征,同时应包含产品名或商标名
应提供器械的一般描述,包括器械的预期用途和预期用户。
参照前一代和相似代际的器械
第2部分:由制造商提供的信息
标签和使用说明书
第3部分:设计与生产信息
材料和组件
系统概述
生产制造信息
参与设计和生产活动的工厂
第4部分:一般安全性和性能要求(GSPRs)
证明符合GSPRs
产品与设计规范
化学、物理和生物学特性
预期连接到其他器械才能正常工作的器械
带测量功能的器械
辐射防护
软件
电气安全性和电磁兼容性
机械风险和热风险防护
第5部分:受益与风险分析和风险管理
风险管理
第6部分:产品验证与确认
样本类型
性能评价与临床证据
此处应包括可证明以下内容的研究:
分析灵敏度
分析特异性
真实性(偏差)
精确性(可重复性和可再现性)
准确性(分析和临床)
检测限和定量
线性度
化验截断
样本处理
干扰物质(内生性和外生性)
交叉反应
上市后监管和上市后性能跟踪:
EURL产品验证
第7部分:稳定性
稳定性包括货架有效期
包装和运输验证
灭菌处理
—致性声明
特定情况中所需的额外信息
伴随诊断
二、IVDR管理体系要求
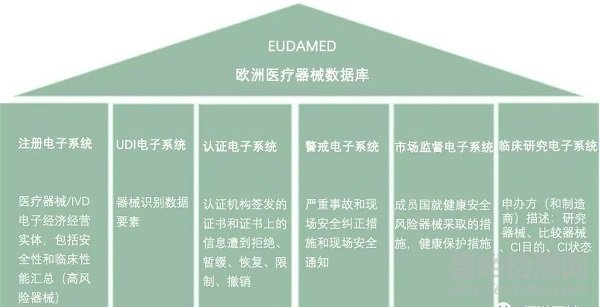
1.IVD将按照风险高低分为4类

2.管理体系过程图示例
3.IVDR管理体系
IVDR是客观看待QMS的一次难得的机会,能确保质量管理体系符合商业目标,同时实现合规性的全部必须要件。
具体来说,在IVDR法规之下,QMS必须至少包含:
法规合规性策略
适用一般安全性和性能要求的标识
管理责任
资源管理,包括供应商和分包商的选择与控制
风险管理
性能评价,包括PMPF
产品实现,包括计划制定、设计、开发、生产和服务提供
验证UDI分配
设置、部署并维护执行一整套上市后监管系统
负责与主管部门、认证机构、其他经济经营实体、客户和/或其他利益参与方进行良好的沟通
警戒管理之下严重事故和现场安全纠正措施的报告流程
纠正与预防措施的管理及其有效性验证
输出、数据分析和产品改善的监督和测量流程
切记,QMS已落实到位,可支持商业机构实现其目标而不是支持其作为实体独立存在。
三、IVDR上市后监管要求
根据第56条和附录XIII的规定开展的性能评价和上市后监管性能评价,包括新的上市后性能跟踪(PMPF)计划必须在QMS当中予以明确定义。这是存在差异变化的另外一个领域,需以器械分类为基础并且应当予以相应的调整适应。因此,QMS流程需要有充分的描述信息,同时允许必要的变化来适应制造商不同的器械类型。PMPF将成为上市后监管活动的关键要素。计划可以是独立文件,或者在整个产品系列当中都保持充分同质性,可以在QMS当中作为一项操作程序或者一套模板为各产品填写完成。如果预期作为程序使用,则程序的范围还需要充分明确地将其识别为计划。计划的引入是较为强烈的指征之一,表明IVDR正在从响应性的PMS(仅依赖于警戒活动)转变为更加主动的体系。上市后监管活动必须根据第78条以及IVDR中的规定予以确立、执行和维护,而且必须作为QMS不可分割的组成部分(见图3)。
尽管上市后监管活动的详细信息并非本文的主题内容,但仍必须注意该流程和QMS的其他领域之间存在诸多的联系,在法律法规中已有相关规定,包括风险管理、性能评价、技术文件更新、纠正和预防措施(CAPA)、不良事件报告和过程/产品监控等等。对于C类和D类的IVD,制造商应制作定期安全性更新报告(PSUR)。
制造商最终还需要利用已有的通用规范(CS)或者“先进性”对比来证实产品符合安全性和性能的全部要求。
四、IVDR欧代和注册要求
Actors模块是第一个需要填写的模块。每个行为人必须输入明确的数据,如姓名、地址、网站等。此外,识别和联系法规符合性人员的详细信息需要与行为人的数据一起输入。非欧洲制造商也必须输入此数据,因此必须向其所在成员国的主管当局申请SRN,并在那里注册其LUA。他们的AR需要先在Eudamed注册。对于初始注册,无需输入AR代表的非欧洲制造商的名称。下一步,非欧洲制造商在其注册过程中指定AR,之后AR验证该注册过程。
SRN-单一注册号
LUA-本地用户管理员
五、IVDR合规建议方案
1.找到合适的分类规则
2.对器械进行合理的分类

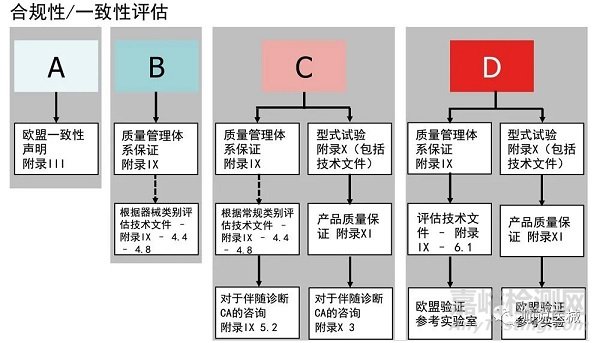
3.合格评定模式选择
4.基于自身产品情况寻找合适的顾问公司和转换时间。