摘 要 Abstract
由于制药行业对专利保护具有高度依赖性,而新药研发的高成本又与公众的用药需求相冲突,为解决这个问题,药品专利链接制度应运而生。美国Hatch-Waxman 法案作为该制度的起源,对美国乃至全世界的制药行业均具有深远的影响,其通过仿制药简化申请、专利声明、专利挑战等制度保障了仿制药产业的发展,同时也通过专利延长和试验数据保护等方式支持了原研药的创新。
Due to the high dependence of pharmaceutical industry on patent protection and the conflicting nature of high cost of new drug research with public demand for medication, the Pharmaceutical Patent Linkage System has emerged as a solution to solve the increasingly serious issue. The Hatch-Waxman Act in the United States, as the origin of this system, has had far-reaching influence on the pharmaceutical industry both in the United States and the world. It ensures the development of the generic drug through mechanisms such as abbreviated generic drug applications, patent declarations, patent challenges, and so on. At the same time, it also helps support new drug innovation through patent extensions and test data protection.
关键词 Key words
药品创新;仿制药;药品专利链接;Hatch-Waxman 法案
drug innovation; generic drug; Pharmaceutical Patent Linkage; Hatch-Waxman Act
药品是治疗疾病、挽救生命不可或缺的必需品。公众对药品的两项基本需求,一是要“有好药”,二是要“吃得起”。“有好药”取决于制药行业的技术创新,但创新药(原研药)研发具有投资大、风险大、难度高、周期长且创新成果易被复制和模仿的特点,需要通过加强专利保护、维护新药的市场独占利益等方式保证创新药的市场回报,即通过经济刺激鼓励药品的研发和创新。而创新药通过专利保护获得市场垄断地位后,往往价格较高,令诸多患者望而却步,仍然难以获得治疗机会。因此,“吃得起”变得至关重要,需要有更多质量、疗效与原研药一致但价格却相对低廉的仿制药进入市场参与竞争,在法律允许的范围内通过打破原研药对市场的垄断以降低药品价格,从而缓解医保负担,降低用药成本。
由此可见,只有将“有好药”与“吃得起”二者有机结合,才能实现药品的可及性。实现药品的可及性首先需要平衡原研药与仿制药的关系,而药品专利链接制度在药品审评审批过程中对平衡二者关系发挥了重要作用。该制度将药品监管机构、专利机构和法院职能进行互动衔接,在药品审评审批过程中提前梳理潜在的药品专利风险;将仿制药上市许可与原研药专利关系和原研药专利状态相链接,保护药品创新的同时鼓励和促进仿制药尽快上市,进而促进整个医药行业的良性、有序发展。
药品专利链接制度起源于美国的Hatch-Waxman 法案, 即《药品价格竞争与专利期补偿法案》(Drug Price Competition and Patent Term Restoration Act)[1],由美国参议员Orrin Hatch 和众议员Henry A.Waxman 于1984 年联合提出。法案的主要功能就是平衡原研药企业与仿制药企业的利益,在保障原研药专利权的同时,通过加快仿制药上市来鼓励药品价格竞争。在该法案实施之前,仿制药的上市许可程序与原研药一致,必须通过一系列的临床试验来证明自身的安全性和有效性。此外,仿制药企业只能在原研药的专利到期之后,才能利用原研药的数据和信息从事仿制研究和试验,这无疑增加了仿制药的上市难度,同时也变相增加了原研药的市场独占期。而Hatch-Waxman 法案则通过对仿制药施行简化申请、Bolar 例外、专利挑战等制度加快了其上市速度;同时也通过专利延长和试验数据保护等方式加强了对原研药的保护。
在Hatch-Waxman 法案之后, 美国又通过了一系列法案和制度[2-5],最终形成了目前的美国药品专利链接制度。当前,已有加拿大、韩国、日本等国家在参考借鉴美国药品专利链接制度的基础上[6],结合本国的药品产业发展情况建立了相应制度。
2017 年,我国明确提出探索建立药品专利链接制度[7]。随着2021 年6 月,第四次修改的《中华人民共和国专利法》的正式实施,最高人民法院、国家药品监督管理局、国家知识产权局陆续发布配套制度,《药品专利纠纷早期解决机制实施办法(试行)》于2021 年7 月4 日正式发布,中国药品专利链接制度正式建立,既借鉴吸纳了国际经验,也具有相当程度的中国特色。
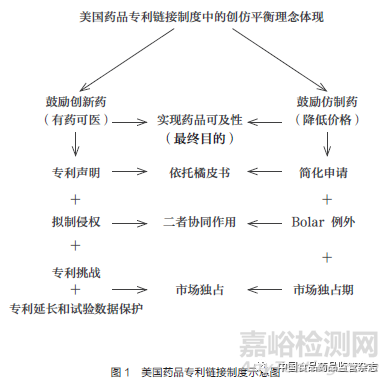
本文将以美国药品专利链接制度作为切入点,通过探析药品专利链接制度的基本结构,让读者对该制度形成初步的认知和了解。首先,通过右侧的简略图示(图1),直观地感受一下美国药品专利链接制度的基本架构和深植其中的创仿平衡理念。
一、橘皮书
橘皮书即《经过治疗一致性评价批准的药品》(Approved Drug Products with Therapeutic Equivalence Evaluation)[8],因封面为橘色,业内惯称为“橘皮书”。橘皮书中收录的是美国食品药品监督管理局(FDA)根据美国《联邦食品药品和化妆品法案》(Federal Food, Drug, and Cosmetic Act)[9],在安全性和有效性基础上批准的药品及其相关专利与专营期信息。橘皮书制度是药品专利链接制度的基础,其核心价值与功能在于药品的信息公开。
原研药企业在申请新药上市许可时,需要提交与该药有关的信息和专利,并通过橘皮书向社会公布,橘皮书中收录的药品信息和专利将作为仿制药的参比对照,为仿制药的研发和申请提供了“标杆”。在仿制药申请阶段,仿制药申请人应对照橘皮书中收录的相关药品信息提出不同类型的专利声明。同时,对于未列入橘皮书的专利,原研药企业也不得在药品专利链接程序中主张权利。
橘皮书于1980 年10 月发布第一版,每年会通过年刊的方式更新过去一年内美国FDA 批准的药品及其专利和专营期信息。除此之外,还会通过增刊的方式按月更新药品信息。橘皮书现有纸质版和网络版两种形式,便于药品行业相关主体查询检索药品信息。
二、仿制药简化申请
创新药的上市,需要经历候选新药研发、临床前研究、临床试验、新药审评审批与上市后监测等复杂过程,尤其是在新药申请前需要通过大量的研究试验形成完整、充分的安全性和有效性数据,整个过程往往需要耗费10 余年的时间并投入巨额的资金。而仿制药的开发则是建立在原研药成熟的技术基础上[10],仿制药申请人只需要证明仿制药与原研药具有治疗等效性即可。
关于仿制药与原研药的治疗等效性,需要满足两项条件,一是药学等效性,即仿制药与参比制剂(reference listeddrug,RLD)具有相同的活性成分、剂型、规格、给药途径和适应症;二是生物等效性,即在相同的实验条件下,仿制药与对照标准制剂(reference standard,RS)在活性成分的吸收程度和速度方面是一致的(差异符合设定标准)[11]。对于仿制药来说,同时满足药学等效性和生物等效性条件,就可以推定仿制药与原研药具有治疗等效性,从而批准仿制药的上市许可。这相比原研药需要取得完整的安全性和有效性试验数据的要求节省了大量的时间与经济成本。
三、专利声明
在仿制药简化申请程序中,针对不同的专利情况,仿制药申请人应对照橘皮书向美国FDA 提出4 种类型的声明:①橘皮书中没有收录相关专利;②橘皮书中收录的相关专利已过期;③承诺在橘皮书中收录的相关药品专利过期前不要求批准所申请仿制药上市并注明专利的过期时间;④橘皮书中收录的相关专利是无效的或仿制药不侵犯相关专利。
原研药企业为了保护自身药品的市场地位,一般会对其药品设立多重的专利保护,在新分子实体或者新化学实体专利的基础上研究制备方法专利、用途专利、剂型专利,组成专利集合并以此成为垄断市场的“武器”,对其他药企形成专利壁垒。但是,在原研药的众多专利中,可能只有一部分是真正具有新颖性、创造性和实用性的产品,其余则是为了阻却竞争对手而刻意设置的技术性障碍,本身不具有科学价值。而专利声明制度,则给了仿制药企业打破原研药企业不当专利禁锢的机会。此外,由于专利的申报瑕疵或者橘皮书的更新滞后,也会导致部分无效的药品专利继续阻却仿制药的上市。因此,专利声明制度设置了上述4 类声明内容,仿制药申请人在进行药品上市许可申请时,需要择一提交。对于提交4 类声明的仿制药申请人将产生以下3 种不同的处理结果。一是提交第①项和第②项声明的,经确认声明属实并通过技术审评后,仿制药即可获得上市许可;二是提交第③项声明的,对于通过技术审评的仿制药,将在原研药相关专利到期后方可获得上市许可;三是提交第④项声明的,仿制药申请人应当在提交申请后20 天内通知原研药权利人(包括药品专利权人和上市许可持有人)并对事实和法律依据进行说明,能否获得上市许可,除了技术审评结论外,还将根据后续诉讼结果处理,该程序即为“专利挑战”程序,也是药品专利链接制度的核心内容。
四、专利挑战
如前文所述,在仿制药申请人提交上述第①至③项声明时,通过技术审评或同时经过专利期限后仿制药即可获得上市许可,这3 种情形属于仿制药上市的常规情形,并不启动专利挑战程序。如果仿制药申请人提交了第④项带有对抗性的声明,则意味着启动了专利挑战程序。该程序要求仿制药申请人应当在提交含有第④项声明的申请后20 日内通知原研药的权利人,并向其说明相关专利无效或者不侵犯相关专利的事实和法律依据,原研药权利人可在收到通知后45 天内提起专利侵权诉讼。
如果原研药权利人提起专利侵权诉讼,美国FDA 将启动30 个月的批准等待期(也称为遏制期或停滞期),该期限内不停止仿制药的技术审评,也不批准仿制药的上市许可,用于等待专利侵权诉讼结果以确认仿制药与原研药之间的专利关系。如在批准等待期内法院作出仿制药侵犯原研药专利权的裁决,仿制药需要等待原研药专利到期后才能获得上市许可;如法院作出不侵犯专利权的裁决,通过技术审评的仿制药将获得上市许可;如法院未能在此期限内作出生效裁决,美国FDA可对通过技术审评的仿制药颁发临时上市许可,上市后的侵权风险由仿制药申请人自行承担;如仿制药申请人与原研药权利人在此期限内达成和解,美国FDA 可根据和解内容和技术审评结果作出是否许可上市的决定,但和解方应将和解协议提交至联邦贸易委员会和司法部进行反垄断审查,主要目的是避免仿制药企业与原研药企业通过达成反向支付协议等方式损害社会公众及其他市场主体利益;如在此期限内原研药的相关药品专利到期,仿制药申请人可以将其第④项声明变更为第②项,通过技术审评的仿制药将获得上市许可。
如果原研药权利人没有在45 天内提起诉讼,美国FDA 可直接根据技术审评结果作出是否批准仿制药上市的决定。需要注意的是,即使原研药权利人没有在该45 天内提起诉讼,也不妨碍其在仿制药上市后提起专利侵权诉讼,原研药权利人并不因此丧失其维权权利,其丧失的是30 个月的遏制期利益,因为在遏制期内原研药实际上仍然享受着市场独占利益。此外,一旦仿制药提前上市,则会对原研药构成市场竞争,可能会导致原研药价格大幅下跌,产生“专利悬崖”现象[12],即使通过日后的专利侵权诉讼也难以挽回其损失,因此绝大部分原研药权利人都会在45 天内积极提起诉讼。
五、市场独占期
为了鼓励仿制药尽早上市,激发仿制药企业挑战原研药专利的积极性,对于首个提交第④项声明进行专利挑战并取得成功的仿制药(称之为“首仿药”),自其上市销售之日起或者法院作出生效裁决之日起(以较早时间为准),美国FDA 将给予其180 天的市场独占期,在此期限内美国FDA 不再批准其他仿制药上市。
该制度对于仿制药的研发与申请具有非常积极的意义,虽然仅规定了近半年的市场独占期,但如果首仿药申请人能够充分利用市场独占期,除了有助于收回研发成本外,还能在其他仿制药大量上市之前确立其市场地位。
需要注意的是,如果仿制药没有在以下期限内上市,将被剥夺市场独占期资格。①被美国FDA 批准上市许可后75 天内;②自提交申请后30 个月内;③法院判决、双方和解或原研药专利被撤销后75 天内。具体截止日期的计算方式为:先在①、②项之间取时间较早者,再在时间较早者与③之间取时间较晚者。市场独占期制度的目的在于鼓励仿制药上市,如果仿制药不把握时机尽早上市,自然也就失去了给予其独占地位的意义。并且如前文所述,首仿药申请人如果与原研药权利人达成反向支付协议,其获得市场独占期后会阻却其他仿制药上市,因此有必要对其施加这种失效限制。
六、Bolar 例外
为了实现仿制药的尽早上市,仿制药企业往往需要在原研药的专利到期前就启动仿制研发工作,因为开展药学等效性和生物等效性试验等工作都需要使用原研药的相关专利,这种使用方式虽然在仿制药上市前并不会对原研药的市场份额造成实质性影响,但显然是带有其商业目的。依据美国的专利法,不能认定该涉及原研药专利的使用行为属于不视为侵权的合理使用范畴。相反,如果要求仿制药只能在原研药专利到期后再开展仿制研究,在经过相对较长的研发试验和审评审批期间后仿制药才能实际上市,这无疑变相延长了原研药的专利保护期。因此,需要在立法层面明确这种使用原研药专利研制仿制药的行为不属于侵权行为,由此就诞生了Bolar 例外制度[13]。
简言之,Bolar 例外就是为了豁免仿制药开发过程中的专利侵权风险,通过制度明确在原研药专利到期前,仿制药企业及相关主体为了向美国FDA 提交仿制药申请资料而进口、制造、使用专利药品进行试验,以获取相关数据信息的行为,即使落入了原研药的专利保护范围也不构成侵权。该制度起源于美国Bolar 公司的一起仿制药研究行为[14],属于专利权保护的例外情形,因此名为“Bolar 例外”。
Bolar 例外制度平衡了原研药的专利保护需求和仿制药的尽快上市需求,缩短了仿制药的上市迟延,使公众可以在原研药专利到期后,尽早获得价格相对低廉的仿制药。
七、拟制侵权
对于仿制药企业,其需要在仿制药上市之前厘清专利风险,否则不但其仿制药可能面临退市风险,还有可能承担高额的侵权赔偿。对于原研药企业,前文中也曾提到,仿制药一旦上市,原研药可能面临“专利悬崖”效应,即使通过日后的专利侵权诉讼也难以挽回其市场损失。因此,为了前置性地解决仿制药的专利侵权问题,使可能发生的专利侵权风险在仿制药上市前得以解决,以此给予原研药和仿制药更多的市场确定性,美国药品专利链接制度创设性地运用了拟制侵权制度,即任何人在他人的药品专利保护期内向美国FDA 提出仿制药注册申请的,均构成侵犯专利权。这使得仿制药的注册申请行为具有可诉性,为专利挑战中的法院诉讼环节提供了制度基础,原研药专利权人通过提前诉讼能及时制止未来可能发生的侵权行为,而暂不考虑实际侵权和损害情形。
需要说明的是,这种人为赋予原研药专利权人的诉讼权利仅是一种程序性权利,其不会因此诉讼而获得实质性赔偿,因为仿制药尚未上市,实质性损害并没有产生,如果确认仿制药侵犯了原研药的专利权,原研药专利权人能够获得的仅是一种程序性禁令,即在原研药专利保护期满前仿制药不得获批上市。
从表面上来看,Bolar 例外与拟制侵权是彼此矛盾的,一个规定不视为侵权,而另一个规定构成侵权,实际上正是这两种制度的有机结合才使得仿制药的研发与申请能够有效推进,同时还能提前规避专利侵权风险。在仿制药研发阶段,通过Bolar 例外对其研发行为进行赋权;在仿制药申请阶段,通过拟制侵权让原研药专利权人对仿制药的申请上市行为进行诉讼以提前排除专利侵权风险。我们也可以这样理解,仅在申请注册的仿制药技术本身不存在专利侵权情形的前提下,为了开发仿制药而使用原研药相关专利的行为才不构成侵权。
八、专利延长
前几项制度更多的是为了促进仿制药尽早上市,为了达到“创仿平衡”的效果,对原研药因临床试验和审评审批延误的上市时间,在专利期限届满后给予其适当延长,也有利于鼓励原研药企业对于新药研发的积极性。在新药上市过程中,很多专利往往在新药研发阶段就需要立即申请专利保护,而此后的临床试验和新药审评审批环节往往又要耗费大量的时间,很多新药在上市时其相关专利的保护期已所剩无几,因此有必要对其进行额外的延长保护以保证其市场回报。
专利延长需要由专利权人在美国FDA 批准上市许可后60 天内向美国专利及商标局提出申请,专利延长的适用范围包括产品专利、制备方法专利和医药用途专利,具体延长时间为临床试验时间的一半与审评时间之和,最长为5 年,但从药品上市之日起算专利保护的总期限不超过14 年。为了避免专利延长制度的滥用,同时还规定对于同一种药品包含多个专利的,只能选择申请其中1 项专利给予延长,而且对于专利权人没有尽到合理注意义务所导致的期限延误不予保护。
九、试验数据保护期
与药品专利延长制度相同,药品试验数据保护期也是给予原研药权利人的一项激励措施。试验数据是原研药企业在药品研发过程中为了进行新药的开发以及证明新药安全性和有效性所形成的一系列研究数据,主要包括临床前试验数据和临床试验数据。试验数据保护是区别于传统的知识产权保护,也有别于一般性商业秘密保护的一种特殊保护制度,其通过给予试验数据权利人一定期限的数据独占权,来阻止仿制药申请人使用其数据获得上市许可,同时也禁止美国FDA 运用试验数据对仿制药进行审评审批[15]。
即使原研药的专利已经到期,在试验数据保护期内,仿制药申请人也不得利用其试验数据进行仿制药申请,而仿制药申请人自行取得试验数据的情形除外。由此可见,试验数据保护是独立于药品专利保护的另一种保护制度,其目的也是为了在一定期限内限制仿制药、me-too 药和me-better 药的搭便车行为,鼓励原研药的开发。
享受试验数据保护的前提是该试验数据所支持的药品已经获得上市许可且试验数据本身尚未被披露。具体的数据保护期从该药品获得美国FDA 批准之日起计算,其中含有新化学实体的新药享有5 年的数据保护期;罕见病药享有7 年的数据保护期;具有新适应症、新剂型或新给药途径的已获批药品享有3年的数据保护期;儿科用药在前述药品的原保护期基础上增加半年;抗生素享有10 年的数据保护期;新生物制品享有12 年的数据保护期。
以上为美国药品专利链接制度的主要构成,其通过Bolar 例外、仿制药简化申请、首仿药180 天市场独占期等制度促进了仿制药的上市,同时还通过延长专利保护期和试验数据保护制度提高了原研药企业的收益预期。药品专利链接制度的核心价值在于通过制度设计实现原研药企业与仿制药企业之间的利益平衡,既鼓励新药研发,也促进仿制药尽早上市,构建创仿平衡的药品市场格局,利用原研药企业和仿制药企业追逐自身经济效益的动能来满足社会公众的用药需求,以此实现药品的真正可及。