警告信是FDA 对被检查企业或个人存在违规行为而采取的监管措施。该文对2018 至2021 年FDA 发给中国药品生产企业的药品cGMP 警告信缺陷项目进行统计,并与国内文献研究中我国药品GMP 检查缺陷情况进行对比,分析我国药品生产企业普遍存在的缺陷以及FDA 和我国药品监管机构在药品GMP 检查重点方面的区别,为我国药品生产企业改进缺陷项目和通过药品GMP 检查提供建议。
美国FDA 的药品检查工作主要由下属的药品审评与研究中心(Center for Drug Evaluation and Research,CDER) 和监管事务办公室(Office of Regulatory Affairs,ORA) 负责开展[1]。警告信是FDA 针对现场检查中发现的违规行为采取的主要监管措施,警告信中会明确指出被检查者的具体违规行为以及违反的相关法律条款,并要求其在期限内做出回复与整改,若违规者没有认真执行,FDA 将会作出更为严厉的监管措施,企业的声誉也会受到严重影响[2—3]。通过对警告信缺陷内容的分析,可以了解现行良好生产质量管理规范(Current Good Manufacturing Practice,cGMP) 检查的重点以及企业在药品生产活动中的薄弱环节,以期为我国药品生产活动和药品GMP 检查提供借鉴。
1、FDA 发给中国药品生产企业cGMP 警告信的统计分析
1.1数据来源
从FDA 官方网站上(https://www.fda.gov/) 查找2018 年1 月至2021 年12 月发布的警告信,筛选出FDA 向中国药品生产企业发布的与药物制剂有关的cGMP 警告信( 以下简称“警告信”),对警告信的数量、缺陷内容进行统计分析[4]。
1.2年份与数量情况
在2014 至2018 年,FDA 每年向中国药品生产企业发出的警告信数量呈增长趋势;而在2018至2021 年,FDA 共计发给中国药品生产企业35 封警告信,年度警告信数量呈下降趋势。其中,2018年15 封,2019 年降至10 封,这可能与缬沙坦等药品安全事件发生后,国内药品生产企业加大了质量管理力度有关。2020 和2021 年,新型冠状病毒病的流行,部分药品生产企业生产活动暂停,同时cGMP 现场检查工作难度加大,导致警告信数下降至每年5 封。上述数据显示出近年来我国对药品监管工作的重视和取得的进步[5]。
1.3缺陷章节分布及逐年变化情况
对纳入本研究的35 封警告信的缺陷项目进行统计,并按照《美国联邦法规》第21 篇211 部分( 以下简称“21 CFR 211”) 的章节进行归纳,发现累计缺陷项目频次为133 次,共涉及21 CFR 211 的8个章节。
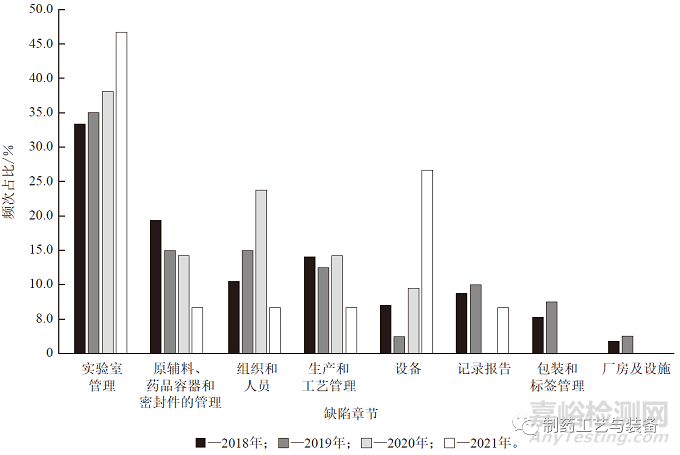
21 CFR 211 共包括11 个章节,警告信中表述的缺陷项目出现在除总则、仓贮和销售、退回和回收的药品3 个章节外的其他各章节中。其中,归类在实验室管理章节的缺陷项目出现频次最高,为48次,占全部缺陷项目频次的36.1% ;其次是归类在原辅料、药品容器和密封件的管理,组织和人员,以及生产和工艺管理3 个章节中的缺陷项目,占比依次为15.8%、13.5%、12.8% ;归类在设备、记录报告以及包装和标签管理3 个章节中的缺陷项目较低,占比依次为8.3%、7.5%、4.5% ;归类在厂房及设施章节中的缺陷项目最少,仅为1.5%。
2018 至2021 年警告信中缺陷项目归属的章节变化情况如图1 所示。2018 至2020 年,归类在实验室管理、组织和人员章节中的缺陷项目频次占比逐年上升,归类在原辅料、药品容器和密封件的管理章节的缺陷项目逐年下降,归类在生产和工艺管理章节中的缺陷项目占比维持在13.0%左右。2021年,除归类在实验室管理和设备2 个章节中的缺陷项目占比大幅增长外,其他章节均呈现下降趋势甚至无缺陷项目。
图1 2018 至2021 年cGMP 警告信的缺陷项目归类及占比情况
归类在实验室管理章节中的缺陷项目占比一直居于首位且呈上升趋势,可见实验室管理一直是药品生产企业的常见问题,是监管的薄弱环节,并且目前仍未得到改善。2021 年,归类在设备章节的缺陷项目占比明显增加,达到26.7%。这可能是2021年疫情防控常态化管理后药品生产企业恢复生产,但未能及时对生产环境和设备仪器进行检测和维修,导致设备方面的缺陷问题增加。尽管归类在组织和人员章节中的缺陷项目占比在2021 年明显下降,但前3 年一直保持增长趋势,在2020 年甚至达到23.8%。所以企业仍需不断加强对质量保证(QA)部门的监管,提高工作人员的工作意识及水平。
1.4缺陷项目统计分析
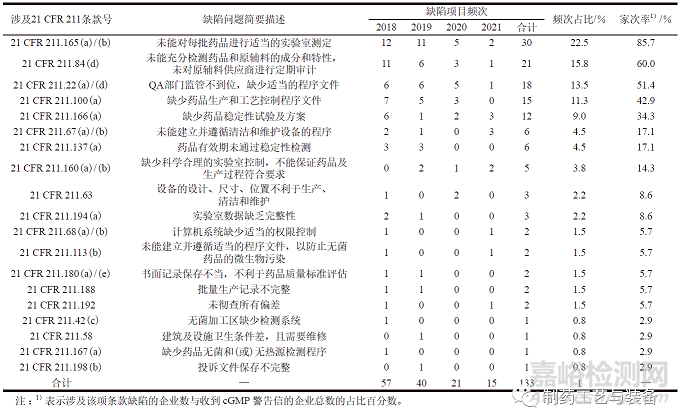
2018 至2021 年,FDA 对中国药品生产企业发出的35 封cGMP 警告信中,累计缺陷项目涉及21CFR 211 中19 项条款,每年缺陷项目涉及21 CFR211 条款情况以及主要缺陷问题如表1 所示。
表1 2018 至2021 年cGMP 警告信中缺陷项目涉及21 CFR 211 条款的情况
1.4.1 药品检验缺陷
如表1 所示,与药品生产过程中药品检验缺陷相关的条款有5 项:21 CFR 211.165(a) /(b)、21 CFR 211.84(d)、21 CFR 211.166(a)、21 CFR211.136(a)、21 CFR 211.167(a),累计缺陷频次占比52.6%,其中最突出的是21 CFR 211.165(a)/(b)(未能对每批药品进行适当的实验室测定),家次率高达85.7%。警告信中指出药品生产企业在药品出厂放行前未能进行适当的实验室测定,未达到对成品药物进行关键的质量控制检测,以确保药品符合最终规格的要求。此外,药品检测缺陷中还存在以下问题:①生产企业未能验证每种药品成分的特性;②缺少或未通过药品稳定性检测,无法为药品的贮存条件和有效期提供数据支撑;③未对每批无菌和(或) 无热原药品进行适当的质量检测,例如在未进行无菌检测的情况下,出厂多批无菌药品。
1.4.2 质量管理程序缺陷
质量管理包括制定质量方针和质量目标以及质量策划、质量控制、质量保证和质量改进[6]。缺陷项目涉及的19 项条款中,有7 项条款与质量管理有关, 包括21 CFR 211.22(a) /(d)、21 CFR211.100(a)、21 CFR 211.160(a) /(b)、21 CFR211.68(a) /(b)、21 CFR 211.113(b)、21 CFR211.192、21 CFR 211.42(c),累计缺陷频次占比为33.9%。
质量管理程序缺陷主要分为以下3 个方面。一是缺少程序文件:①企业未能提供QA 部门履职程序文件,QA 不能有效履行相应职责;②未能制定药品生产和工艺控制程序,以确保药品的特性、质量等;③未能制定科学、合理、明确的实验室控制标准,不能确保正确的实验室操作;④未能建立无菌、灭菌验证过程的程序文件,不能确保无菌药品符合标准。二是缺少控制系统或权限:①企业未能对计算机或相关系统进行适当控制,缺少访问权限,例如药品分析实验室的实验员都能够以“系统管理员”的身份登录系统,不需要密码,并且具有完整的系统管理权限;②无菌加工区缺少检测系统,不能保证数据的完整性和可靠性。三是对于出现的偏差和不合格结果,未进行彻底调查。
1.4.3 数据完整性缺陷
数据完整性是指贯穿整个数据生命周期的数据采集的完整、一致和准确的程度,是保证药品生产企业有效开展生产活动的基础[7]。由表1 的缺陷情况可见,数据完整性问题主要源于2 方面。一是数据记录不完整:①生产记录不完整,如未列出所有药品成分、缺少试验的关键参数、试验记录表中只保留最终结果而未保留试验过程中的数据等;②存在篡改、伪造数据等造假行为。二是记录、文件的保存不当,如生产记录、投诉文件等保存不当,造成遗失、缺损等问题,不利于药品质量评估工作的开展。
1.4.4 环境与设备缺陷
环境与设备是保障药品生产的基础条件,也是影响药品质量和企业效益的重要因素。环境与设备方面的缺陷有:①缺少或未遵守设备清洁和维护的程序文件;②设备的设计、尺寸、位置不利于生产、清洁和维护;③建筑及设施卫生条件差,且需要维修,例如在生产车间的墙面上发现有裂缝、霉菌、虫害,严重影响药品质量。
1.4.5 缺陷项目的逐年变化情况
2018 至2021 年, 缺陷项目集中在21 CFR211.165(a) /(b) (药品放行前的质量标准检验)、21 CFR 211.84( d) ( 样本的检查和测试)、21 CFR211.22(a)/(d)(QA 部门履职及程序文件)、21 CFR211.100(a) (生产及工艺控制程序文件)4 项条款,且整体上呈现下降趋势。而21 CFR 211.166(a)(稳定性试验及方案)、21 CFR 211.67(a)/(b)(设备清洁和维护)、21 CFR 211.160(a)/(b) (实验室控制和科学合理性) 的频次占比大幅上升。近2年,实验室药品检测、程序文件的制定与遵守、数据完整性等高频次缺陷条目方面有了明显改善。FDA 在2016 年发布了《数据完整性和cGMP 合规行业指南草案》,数据可靠性等问题越来越受到企业重视。随着药品监管力度的不断加强,这些高频次缺陷依然是企业在生产过程中常见的风险点。同时,环境与设备、无菌药品检测、药品稳定性检测程序文件等项目中一些低频次缺陷的再次出现,提示其不应成为生产管理中的忽略环节,尤其是环境与设备方面,缺陷频次占比增长态势明显。药品生产设备与环境等硬件系统,是决定药品质量的重要因素,仪器、设备、系统的调整与更新,应当紧跟药品质量提升的步伐。
2、与国内药品GMP检查的对比分析
2.1国内药品GMP检查缺陷情况
随着2019 年新修订的《中华人民共和国药品管理法》颁布实施,药品监管的方式发生了重大变革,全面提升了对落实药品生产企业主体责任与加强监管的要求[8]。通过查阅近年来国内关于药品生产企业GMP 检查缺陷分析的文献,发现在针对国内整体以及部分省份的药品GMP 检查研究中,出现缺陷项目次数较多的均归类在质量控制与质量保证章节,如本课题组在对我国2016 至2019 年被收回GMP证书的药品生产企业存在的缺陷研究中,观察到归类在质量控制与质量保证章节的缺陷项目频次较高,占比高达23.3%,其中质量控制实验室管理中的缺陷项目占该章节缺陷的49.8% [9—10]。
其他缺陷项目归类较多的章节包括物料与产品、文件管理、设备、机构与人员、附录(计算机化系统)。张庆芬等[11] 对广东省2018 至2020 年药品生产企业在跟踪检查中发现的缺陷项目进行分析,观察到缺陷项目主要归类在质量控制与质量保证、文件管理、机构与人员等章节,问题主要是检验操作、人员资质与培训、批生产记录不符合要求等。金建闻等[12] 对河南省210 份GMP 认证现场检查报告中的缺陷项目进行统计,观察到缺陷次数归类较多的章节依次为:质量控制与质量保证、文件管理以及设备,分别占缺陷总数的19.7%、15.6%和13.1%,并且对比发现河南省、山东省、辽宁省、安徽省和河北省的检测结果中缺陷分布情况一致。
2.2FDA与国内药品检查缺陷情况对比
对比FDA 发给中国药品生产企业的cGMP 警告信和国内药品GMP 检查的缺陷情况可见,主要缺陷均为质量控制与保证、物料与产品、文件管理、设备与环境、机构与人员这5 个方面,但涉及缺陷的具体内容及关键问题也存在一定差异。
2.2.1 质量控制与保证
质量控制与保证方面的缺陷是FDA 与我国GMP 药品检查的主要缺陷内容,且缺陷频次最高的均为实验室管理部分,包括药品成分、稳定性、无菌检验等实验室检测不充分,缺少科学合理规范的检测程序等。其次,缺少计算机系统权限管理、偏差处理不彻底也是共同缺陷项目,但FDA 只有少数警告信涉及该缺陷项目,而国内GMP 检测在计算机系统权限和偏差处理方面发现缺陷的情况较为常见,如文献[9] 报道计算机系统权限以及偏差处理方面的缺陷是高频次缺陷项目。
2.2.2 物料与产品
在物料与产品方面,FDA 检查缺陷对应的是原辅料、药品容器和密封件的管理章节,主要问题是未能充分检测药品和原辅料的成分、特性,以及未对原辅料供应商进行定期审计;而我国GMP 检查发现的突出问题为物料产品贮存、标识、留样等操作不规范。
2.2.3 文件管理
文件是质量保证系统的基本要素,做好文件管理有助于产品质量追溯[13]。在文件管理方面的缺陷均涉及缺少关键性质量管理程序文件,如记录缺失、造假等数据可靠性问题。不同的是,cGMP 警告信中的突出问题为缺少程序文件,而国内针对药品生产企业数据可靠性进行的研究较多。
2.2.4 设备与环境
设备与环境方面涉及的缺陷内容包括厂房和设备的设计、使用和清洁、维护和维修,cGMP 警告信和国内药品检查通告中均指出了具体的缺陷问题,如缺少操作规程、维护计划,使用和维修记录不完整,生产设备未标明状态标识或标识信息不完整等。该方面的缺陷尽管不是FDA 和国内药品GMP 检查的突出问题,但依然需要药品生产企业生产加以重视。
2.2.5 机构与人员
FDA 警告信中关于机构与人员方面的缺陷问题主要归类在组织和人员章节,包括企业质量管理部门缺少程序文件、质量部门人员未能有效履行职责。此外,有少数警告信中对企业提出要加强工作人员的培训。我国药品GMP 检查中存在的缺陷问题包括人员资质或数量与生产活动不匹配、未能建立完整的培训制度、岗位操作不规范等[14]。
2.3缺陷差异分析
硬件系统、软件系统和人员是药品生产的三要素。通过以上缺陷情况对比可见, FDA 和我国GMP 检查在硬件系统方面发现的缺陷情况相似,而针对软件系统和人员方面的缺陷,突出的问题不相同,这一方面是由于美国与我国有关药品GMP的法律法规存在差异,另一方面是检查对象的不同。
首先,在cGMP 和GMP 规范方面,FDA 对软件系统的要求较多,而我国药品GMP 对硬件系统的要求多。在软件系统方面,我国药品GMP 检查缺陷问题多为生产行为的错误、缺失,属于操作层面;而cGMP 警告信中的缺陷问题多为程序文件缺失,属于管理层面。此外,结合我国GMP 检查和FDA 发布的药品检查员招聘公告,我国对药品检查员的准入有更严格的要求,而FDA 对人员职责的约束更为细致,且培训更加全面。因此,在我国药品GMP 检查缺陷中,人员的履职、培训问题更为突出[15—16]。
其次,在FDA 向我国药品生产企业发出的cGMP 警告信中,检查对象为出口药品,需要符合我国GMP 和cGMP 的检查要求,取得《药品出口销售证明》等文件,并且FDA 对进口药品的登记和审批、设施注册以及原料药药品主文件(drug master file,DMF)备案等都有着明确的制度要求[17]。国内药品检查主要依据GMP 规范,接受常态化、不定期的药品GMP 符合性检查和监督检查。由此可见,出口药品需要接受中、美两方的检查,检查程序更多,检查内容也更全面。
3、建议
3.1制定程序文件,提升软件水平
完善的质量管理体系对于保障药品质量以及生产有序运行起到关键性的作用,而程序文件是质量管理体系不可或缺的基本部分,是员工开展工作的规范和依据[14]。通过对cGMP 警告信和国内药品GMP 检查情况的分析,程序文件的缺失是药品生产企业在软件系统中的突出问题。企业需制定并完善药品生产和工艺控制、设备清洁和维护、质量部门履职程序等环节的关键性质量管理程序文件及书面计划,且文件需符合各部门的职责与程序,具有规范性、系统性和可操作性,确保部门工作人员能够充分了解并掌握工作要求、有效履行职责、促进质量管理体系的运行,从而提升企业的软件水平。
3.2加强人员培训,健全培训体系
人是实施药品GMP 最核心、最积极的要素,是药品生产企业实施质量管理活动的基础[18]。国内药品检查多次指出要加强人员培训,cGMP 警告信中也曾要求生产企业在回复时提供员工培训计划,因此企业必须建立完善的培训体系,制定切实可行的培训计划,针对不同岗位、不同层次的人员开展培训。另外,培训过程中要注重理论与实践相结合,不断创新培训方式。企业对员工进行培训后,需及时对培训结果进行考核,并将考核结果与奖惩挂钩,杜绝个别员工应付培训的思想[19]。随着药品相关法律法规及GMP 附录的不断更新和完善,企业在培训内容上也要与时俱进,保证培训的数量与质量,不能“一培永逸”。
3.3聚焦高风险问题,增强检查个性化
对cGMP 警告信和国内药品GMP 检查的缺陷分析结果显示,近年来企业在高频次缺陷方面的改善效果并不明显,部分缺陷项目占比甚至出现逐年上升的趋势,因此企业应当注重对高频次缺陷问题的管理。企业在接受检查后,应及时对缺陷问题进行分析、评估,并结合以往检查经验,制定具有针对性的整改措施。在日常生产中,企业可加强对此类风险问题的自检;同时,检查组可基于以往检查情况,对高频次缺陷问题进行重点检查,并根据生产药品特性,对企业实施个性化检查,从而最大限度利用有限的检查资源[20]。
3.4借鉴警告信制度,完善药品GMP检查机制
国家市场监督管理总局于2020 年颁布的《药品生产监督管理办法》中,第五十九条指出“药品存在质量问题或者其他安全隐患的,药品监督管理部门根据监督检查情况,应当发出告诫信,并依据风险相应采取暂停生产、销售、使用、进口等控制措施”[21]。对药品生产中的违规行为发布告诫信,是一种劝导性的监管措施,既能让企业发现生产与管理中的缺陷与不足,又给予自我纠正的机会,有助于药品生产动态监管的实施。FDA 警告信制度贯穿于药品全生命周期的监管,在警告信设计、发布、终止环节,具有相对成熟、缜密的体系。我国药品监督管理部门可结合以往药品GMP 检查经验,借鉴FDA 警告信制度,进一步完善药品GMP 检查机制。
参考文献
[1] 颜若曦, 曹 轶, 董江萍.FDA对我国药品生产企业检查分析[J].中国新药杂志, 2020, 29(15): 1697-1701.
[2] 宋华琳, 刘 炫.美国FDA警告信的制度架构及启示[J].中国食品药品监管, 2019, (12): 28-35.
[3] FDA.Regulatory Procedures Ma n u a l , Chapter4-ADVISORY ACTIONS [EB/OL].(2022-02-18)[2022-08-26].https://www.fda.gov/inspections complianceenforcement-and-criminal-investigations/compliancemanuals/regulatory-procedures-manual.
[4] FDA.Warning Letters.[EB/OL].(2022-08-24)[2022-08-27].https://www.fda.gov/inspections complianceenforcement-and-criminal-investigations/complianceactions-and-activities/warning-letters.
[5] 周清萍, 周 娜, 梁 毅.关于数据管理和数据完整性的药品cGMP警告信分析[J].化工与医药工程, 2020,41(1): 62-68.
[6] 叶 笑, 颜若曦.产品质量回顾的要点分析研究[J].现代药物与临床, 2022, 37(7): 1653-1656.
[7] 张铁军, 韩文涛, 韩 静.数据完整性对中国制药企业GMP检查的影响分析[J].中国新药杂志, 2017, 26(9):985-989.
[8] 王 丹, 欧阳楠, 陈 颖.新法规要求下药品生产检查形势与监管策略探讨[J].中国药事, 2022, 36(6): 611-615.
[9] 曹琳琳, 武志昂.2016—2019年被收回GMP证书的药品生产企业存在的缺陷分析及改进建议[J].中国现代应用药学, 2021, 38(9): 1107-1113.
[10] 曹琳琳, 武志昂.跟踪检查和飞行检查中收回GMP证书的药品生产企业缺陷研究[J].中国医药工业杂志, 2021,52(4): 574-580.
[11] 张庆芬, 招伟汉, 江映珠, 等.广东省药品生产企业GMP跟踪检查情况分析[J].广东化工, 2021, 48(15): 100-101.
[12] 金建闻, 党明安, 谢芝丽.河南省药品GMP认证缺陷分析及新修订《药品管理法》实施后的建议[J].中国药学杂志, 2021, 56(2): 162-166.
[13] 马小燕, 高嘉敏, 谢二磊.2020年江西省药品生产企业检查缺陷项目分析及对策[J].中国药业, 2021, 30(23): 15-18.
[14] 刘学良, 韩达斌, 潘 平, 等.青海省中药饮片生产企业新版药品GMP认证检查缺陷分析及改进建议[J].中国药事, 2016, 30(9): 869-873.
[15] FDAJOBS.Consumer Safety Officer [EB/OL].(2022-08-22)[2022-08-27].ht tps : / /www.usajobs.gov/job/672521800.
[16] 上海市药品监督管理局.国家药品监督管理局关于印发职业化专业化药品检查员分级分类管理办法的通知[EB/OL].(2021-06-18)[2022-08-26].https://yjj.sh.gov.cn/fg-gfxwj/20220301/d02e6f98e9bc4ee591409aaceab0b5df.html.
[17] 柴倩雯, 李晓宇, 尤晓敏, 等.中美药品进口注册管理制度研究[J].中国药学杂志, 2016, 51(9): 772-776.
[18] 钱利武, 罗京京, 王 浩, 等.药品 GMP 检查中质量控制与质量保证方面存在的主要问题及建议[J].中国药事,2020, 34(1): 17-21.
[19] 张涛志, 胡永新, 邵倩倩, 等.广东省撤销GSP证书的药品批发企业缺陷项目分析[J].中国药事, 2021, 35(10):1116-1122.
[20] 杨璐瑶, 杨 悦.美国FDA基于风险的药品检查计划的研究与借鉴[J].中国新药杂志, 2020, 29(22): 2535-2540.
[21] 国家市场监督管理总局.药品生产监督管理办法[EB/OL].(2020-01-22)[2022-08-27].https://www.samr.govcn/zw/zfxxgk/fdzdgknr/fgs/art/2023/art_65070d0ee03a4109ac831ee7b3cee51c.html.
本文作者曹琳琳、钱雅婷、张青松、李海剑、肖皓祥、刘伟,河南省药品审评查验中心、郑州大学药学院,来源于中国医药工业杂志,仅供交流学习。