注射用微球无菌检查既要关注“球外无菌”又要关注“球内无菌”,而检测的难点是“球内无菌”,即微球内部的无菌性检查。该研究在调研注射用微球生产工艺的基础上,选择代表性品种,模拟潜在微生物污染物存在条件下的微球溶解过程,建立了2种内部无菌检查方法:一是采用二甲基亚砜(DMSO) 溶解微球;二是采用pH 10.0缓冲液溶解微球。2种方法制备的无菌检查供试液均以细菌耐受性芽孢作为试验菌,采用直接接种法进行无菌检查,并探讨供试液制备、试验用菌株、方法适用性等关键技术环节的科学性和合理性。该研究是注射用微球无菌检查方法开发和验证策略的有益探索,可为此类药物制剂的质量控制和标准提高提供研究思路和数据支撑。
随着制药工业的发展,为适应疾病的治疗或预防需要而采用的药物剂型层出不穷[1—2]。药物与适宜载体(生物可降解材料) 经过一定的分散包埋技术制得的微米或纳米级固态、液态或气态药物,包括微囊、微球、微晶、脂质体和纳米粒等,称为微粒制剂,其中注射用微球是应用最广泛的微粒制剂之一。从国家药品监督管理局网站上可查询到30余个注射用微球的进口、国产品种的批准文号,代表性品种有注射用醋酸亮丙瑞林微球、注射用利培酮微球、注射用醋酸奥曲肽微球、注射用全氟丁烷微球等。这些品种的质量控制和分析方法备受关注,其中无菌检查法的科学性和合理性是其关注的重要内容。
关于微粒制剂的质量控制,美国药典(USP42)<71>、欧洲药典(EP 11.0)<2.6.1>、日本药典(JP18)<4.06> 等均未单独收载通用性技术要求的章节,仅在制剂通则中对处方设计和制备工艺进行了介绍性说明,未涉及微生物检查方法的特殊规定;美国注射剂协会(PDA) 技术报告、人用药品技术要求国际协调理事会(ICH) 指导原则中也未涉及相关规范性要求;《中华人民共和国药典》2010 年版(ChP2010) 附录首次收载《微球、微囊和脂质体制剂指导原则》,ChP 2015 修订为《微粒制剂指导原则》(通则9014),一直沿用至ChP 2020。通则9014 中对于药物载体类型、常用辅料载体和质量控制的检查项目等进行了规定,但未对微生物质量控制提出明确的技术要求[3]。
目前,注射用微球品种标准中无菌项下规定主要分为3类。
①指向通则,未规定具体操作方法,如:取本品,加所附的助悬剂制成混悬液,依照无菌检查法进行检查,应符合规定。
②指向通则,规定了具体操作方法,如:取本品,每支按说明书,用所附的注射器和针头,加入所附的溶剂使分散均匀,采用直接接种法,取每支全量接种至培养基50 ml中,以金黄色葡萄球菌为阳性对照,依照无菌检查法进行检查,应符合规定。
③规定分别进行微球内部和外部的无菌性检查,如:取本品,分别溶解于相应培养基2 ml 中,过滤,以金黄色葡萄球菌为阳性对照菌,进行外部无菌检查;取本品,分别溶解于无菌二甲基亚砜(DMSO)2 ml 中,过滤,且每膜过滤体积不超过10 ml,以金黄色葡萄球菌为阳性对照菌,进行内部无菌检查。
注射用微球无菌检查的难点聚焦在“球内无菌”,即微球内部的无菌性检查[4]。一般而言,产品采用终端灭菌工艺,且粒度分布小于微生物污染物,生产工艺过程中微生物不易存在或不易存在于制剂内部的,进行微球外部无菌性检查;产品采用无菌生产工艺,但粒度分布大于或类似于微生物污染物,在生产过程可能存在潜在污染风险时,应分别进行微球内部和外部的无菌性检查。
经调研,大部分注射用微球采用无菌生产工艺,以聚乳酸- 羟基乙酸共聚物(PLGA) 作为辅料[5—6],与原料混合后,在密闭系统中包裹形成制剂,生产过程涉及二氯甲烷、乙酸等有机溶剂[7],这种条件不利于微生物的生长和存活,但存在耐受性芽孢的污染风险。代表性品种( 如注射用醋酸奥曲肽微球等) 在进行微球内部无菌检查时,采用DMSO 作为溶剂制备供试液,但高浓度DMSO 具有抑菌作用,是否影响无菌检查方法的适用性和有效性尚待评估。此外,在某些品种( 如注射用利培酮微球) 的方法建立过程中,曾对其生产工艺各环节进行风险评估,认为涉及的无菌操作均在微球硬化之后,潜在的污染物仅可能存在于外表面,故在标准变更时取消了内部无菌检查。上述评估的规范性如何在药品标准中进行通用性技术规定,尚未进行深入探讨。
本研究选择注射用醋酸亮丙瑞林微球、注射用利培酮微球、注射用醋酸奥曲肽微球等国内外代表性品种,聚焦微球内部的无菌性检查,考察供试液制备、试验用菌株、检查方法等关键技术环节的科学性和合理性,以期为注射用微球无菌检查方法的建立和药品标准的制定和修订提供研究思路和数据支撑。
1、仪器与试药
Phenom Nano 型扫描电镜( 荷兰Phenom 公司) ;Supro150 型溅射仪[ 复纳科学仪器(上海) 有限公司] ;MIR254型恒温培养箱(日本Panasonic 公司) ;FOM4/9 型高压蒸汽灭菌器(意大利Fedegari 公司) ;Steritest Equinox 型集菌仪和Milliflex 型薄膜过滤系统均购自德国Merck-Millipore公司。
注射用醋酸亮丙瑞林微球(上海丽珠制药有限公司)、注射用利培酮微球(瑞士Janssen-Cilag 公司)、注射用奥曲肽微球(瑞士Novartis Pharma Schweiz 公司) 和注射用全氟丁烷微球(美国GE HealthCare AS 公司),上述产品均在有效期内。
硫乙醇酸盐流体(FTM) 培养基(批号VM859391903)、胰酪大豆胨液体(TSB) 培养基(批号VM884759920) 和胰酪大豆胨琼脂(TSA) 培养基(批号VM873358915) 均购于德国Merck Millipore 公司;沙氏葡萄糖液体(SDB) 培养基(英国Oxoid 公司,批号2202463);沙氏葡萄糖琼脂(SDA)培养基(批号1086835) 和pH 7.0 无菌氯化钠- 蛋白胨缓冲液(批号1086984) 均购于广东环凯微生物科技有限公司;0.1%无菌蛋白胨水溶液(北京奥博星生物技术有限责任公司,批号20200812);DMSO(国药集团化学试剂有限公司,批号20131225);其他试剂均为分析纯,水为灭菌纯化水。
金黄色葡萄球菌(Staphylococcus aureus)[CMCC(B)26003]、大肠埃希菌(Escherichia coli)[CMCC(B)44102]、铜绿假单胞菌(Pseudomonas aeruginosa)[CMCC(B)10104]、枯草芽孢杆菌(Bacillus subtilis)[CMCC(B)63501]、短小芽孢杆菌(Bacillus pumilus)[CMCC(B)63202]、生孢梭菌(Clostridium sporogenes)[CMCC(B)64941]、白念珠菌(Candida albicans)[CMCC(F)98001]、黑曲霉(Aspergillus niger)[CMCC( F)98003] 等无菌检查用标准菌株均为本实验室保藏。
2、方法与结果
2.1试验菌液的制备
参照ChP 2020 无菌检查法(通则1101),制备大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌、枯草芽孢杆菌、白念珠菌、黑曲霉和生孢梭菌的试验菌液;参照ChP 2020 抗生素微生物检定法(通则1201),制备枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢的试验菌液。分别用pH 7.0 无菌氯化钠- 蛋白胨缓冲液制成每1 ml 含菌数为108 cfu 和不大于100 cfu的菌悬液。
2.2供试液的制备
DMSO 供试液:取注射用微球粉末(装量约为每瓶30 mg),每瓶加入DMSO 溶液2 ml,混匀。另取样品,分别加入10% DMSO、25% DMSO、50% DMSO、75% DMSO 溶液,均无法溶解形成均一的供试液。根据ChP 2020 通则1101 关于供试液接种比例不大于培养基体积10%的规定,后续试验中分别采用100% DMSO( 高浓度)、10%DMSO 溶液(低浓度),考察溶剂对试验菌生长的影响。
水溶性供试液:精密称取氯化铵2.7 g,置500 ml量瓶中, 用水450 ml 溶解, 量取三乙胺6.0 ml加入瓶中,混匀,用水定容,用三乙胺调至pH10.0±0.1,制成pH 10.0 缓冲液,灭菌[8]。取注射用微球粉末(装量约为每瓶30 mg),转移至含上述灭菌缓冲液25 ml 的玻璃瓶中,密封,置45 ℃、120 r/min 的恒温摇床,至微球完全溶解。
2.3方法适用性试验
参照ChP 2020 无菌检查法(通则1101) 配制相应培养基,经培养基的适用性和灵敏度检查,符合药典标准规定。方法适用性试验的设置共分为3组,其中空白对照组用于判定试验方法体系是否正常,溶剂组用于判定溶剂对试验方法是否有干扰,试验组用于判定试验方法是否符合规定。
试验组:取注射用微球,加入相应溶剂(DMSO或pH 10.0 缓冲液),在上述溶液中分别加入含菌量不大于100 cfu 的试验菌液,混匀;将含有大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌和生孢梭菌的混合溶液转移至FTM 培养基50 ml 中,于33 ℃培养不少于5 d,将含有枯草芽孢杆菌、白念珠菌、黑曲霉、枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢的混合溶液转移至TSB 培养基50 ml 中,于23 ℃培养不少于5 d,观察试验菌是否生长。
溶剂对照组:取相应溶剂(DMSO 或pH 10.0缓冲液),加入含菌量不大于100 cfu 的试验菌液,混匀,其他同试验组操作。
空白对照组:取灭菌纯化水,加入含菌量不大于100 cfu 的试验菌液,混匀,其他同试验组操作。
2.4注射用微球的形态考察
分别取少量注射用微球,用小药勺取出少许粉体或粉块,均匀撒至石英玻璃台上,使用一次性刻刀,反复、随机刻切10 次;将上述粉体或粉块均匀撒至样品台导电胶上,使用洗耳球轻吹样品表面,清理未黏附牢固的样品,采用溅射仪(设置溅射功率20 W,镀金时间30 s) 处理,然后装载入扫描电镜(SEM) 仪,根据不同的成像比例,设置加速电压为5 或10 kV。
分别以注射用醋酸亮丙瑞林微球、注射用利培酮微球和注射用醋酸奥曲肽微球为考察对象,其SEM 照片如图1 所示。
A—注射用醋酸亮丙瑞林微球(×2000,加速电压5 kV,比例尺30 μm) ;B—注射用利培酮微球(×2000 倍,加速电压5 kV,
比例尺30 μm) ;C—注射用醋酸奥曲肽微球(×2 000 倍,加速电压10 kV,比例尺30 μm) ;D—注射用醋酸亮丙瑞林微球( 剖面
图,×5000 倍,加速电压10 kV,比例尺10 μm) ;E—注射用利培酮微球(剖面图,×6800 倍,加速电压5 kV,比例尺10 μm) ;
F—注射用醋酸奥曲肽微球(剖面图,×2000 倍,加速电压10 kV,比例尺30 μm)。
图1 注射用微球代表性品种的SEM 照片
结果显示,3 种微球均为实心球体,球面直径为30 ~ 100 μm ;其中注射用醋酸亮丙瑞林微球为蜂窝状实心球体,球体内孔径为1 ~ 3 μm,球面和球内的直径均在微米级,与常见细菌的大小处于同数量级。虽然PLGA 是缓释或控释制剂的常用辅料[6],具有生物可降解性,但在无菌检查供试液的常规制备时间内无法完全溶解。
2.5DMSO 溶解微球后的无菌检查方法适用性试验
2.5.1 DMSO 对试验菌生长抑制作用考察
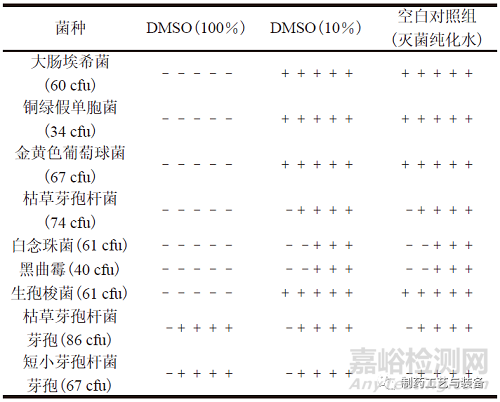
分别取“2.2”项下100% DMSO(高浓度)、10% DMSO 溶液(低浓度) 适量,依照ChP 2020通则1101,分别加入不大于100 cfu 的各试验菌悬液,混匀。将含有大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌和生孢梭菌的混合溶液转移至FTM培养基50 ml 中,于33 ℃培养不少于5 d,将含有枯草芽孢杆菌、白念珠菌、黑曲霉、枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢的混合溶液转移至TSB培养基50 ml 中,于23 ℃培养不少于5 d。结果如表1所示,100% DMSO 具有抑菌作用,药典规定的试验菌(营养体) 均无法生长;枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢可以生长。10% DMSO 溶液无抑菌作用,ChP 2020 规定的试验菌(营养体)和耐受性芽孢均可以生长,但微球无法溶解。
注:1) 每个单元格有5 个正负号,依次显示试验菌接种后d1至d5的
生长情况:“–”表示无菌生长,“+”表示有菌生长。
表1 DMSO 对试验菌生长的抑制作用1)
2.5.2 方法适用性试验
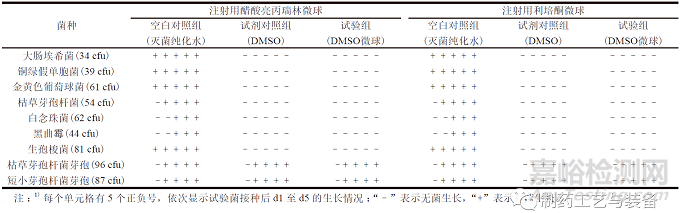
分别选择注射用醋酸亮丙瑞林微球和注射用利培酮微球2个代表性品种,进行微球内部无菌检查的方法适用性试验。取上述样品,每瓶用DMSO溶液2 ml 溶解微球,以此作为供试液。供试液冷冻干燥后镜检,结果显示SEM 视野下未见完整的微球形态,提示辅料已被DMSO 溶液溶解。依照ChP 2020 通则1101,在注射用微球与DMSO 溶液混合后,分别加入不大于100 cfu 的各试验菌悬液,混匀。将含有大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌和生孢梭菌的混合溶液转移至FTM 培养基50 ml 中,于33 ℃培养不少于5 d,将含有枯草芽孢杆菌、白念珠菌、黑曲霉、枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢的混合溶液转移至TSB 培养基50 ml,于23 ℃培养不少于5 d,作为试验组;同时,以灭菌纯化水作为空白对照组、100% DMSO 作为试剂对照组,同法操作。结果如表2所示。
表2 DMSO 溶解注射用微球无菌检查方法的适用性试验结果1)
100% DMSO 可溶解上述2个品种的注射用微球,但“2.5.1”项下结果显示100% DMSO 对于药典规定的试验菌(营养体) 的生长均具有抑制作用,因此可选择枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢作为试验菌,进行方法适用性试验。10% DMSO溶液虽然未见对于药典规定的试验菌(营养体) 的生长抑制作用,但其无法溶解微球制成供试液。结果提示,以上2个注射用微球代表性品种的内部无菌性检查可采用100%DMSO作为溶剂制备供试液,结合生产工艺过程中潜在污染的风险评估,以耐受性芽孢作为试验菌,进行方法适用性试验。
2.6水溶性缓冲液溶解微球后的无菌检查方法适用性试验
根据注射剂的应用情况,无菌检查一般制备水溶性供试液,通过薄膜过滤法进行无菌性检查。DMSO 为有机溶剂,且对营养体状态的试验菌的生长具有抑制作用,为寻求水溶性的“破球”方法,通过模拟注射用微球的体外释放度,制备水溶性供试液。由于PLGA 辅料的酯键断裂后产酸,调节缓冲体系pH 值至碱性,可加速辅料降解[8—9]。
2.6.1 pH 10.0 缓冲液对试验菌生长抑制作用的考察
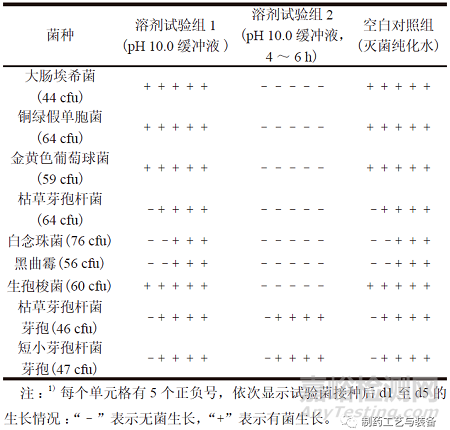
取“2.2”项下pH 10.0 缓冲液适量,依照ChP2020 通则1101,分别加入不大于100 cfu 的各试验菌,混匀。将含有大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌和生孢梭菌的混合溶液转移至FTM培养基50 ml 中,于33 ℃培养不少于5 d ;将含有枯草芽孢杆菌、白念珠菌、黑曲霉、枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢的混合溶液转移至TSB培养基50 ml 中,于23 ℃培养不少于5 d。同时,为确证pH 10.0 缓冲液中试验菌株的稳定性,另取适量pH 10.0 缓冲液,加入试验菌后,置45 ℃、120 r/min 的恒温摇床中孵育4 ~ 6 h,然后分别转移至不同培养基,观察菌株生长情况。将上述采用新鲜配制pH 10.0 缓冲液的试验组作为溶剂试验组1,采用经孵育4 ~ 6 h 的pH 10.0 缓冲液的试验组作为溶剂试验组2。
表3 pH 10.0 缓冲溶液对试验菌生长的抑制作用1)
结果如表3所示,依照ChP 2020 通则1101 的试验步骤,pH 10.0 缓冲液对药典规定的试验菌(营养体)、枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢的生长均无影响;但当温度升高、振荡增加、缓冲液经孵育处理时,试验菌株的生长状态不稳定,仅耐受性芽孢可以生长。
2.6.2 方法适用性试验
分别选择注射用醋酸亮丙瑞林微球和注射用奥曲肽微球2 个代表性品种,进行无菌检查方法适用性试验。取上述样品,每瓶转移至含pH 10.0 缓冲液25 ml 的玻璃瓶中,于45 ℃、120 r/min 密封无菌状态下孵育4 ~ 6 h,获得供试液(同时以pH 4.0、pH 7.0 缓冲液作为对照,相同条件下微球均无法溶解,加速释放行为与相关研究结果一致[9])。供试液冷冻干燥后镜检,SEM 视野下未见完整的微球形态,提示辅料能够在此条件下加速降解。
依照ChP 2020 通则1101,在注射用微球与pH10.0 缓冲液混合后,分别加入不大于100 cfu 的各试验菌悬液,混匀,在上述条件下孵育至微球完全溶解。将含有大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌和生孢梭菌的混合溶液转移至FTM 培养基225 ml 中,于33 ℃培养不少于5 d ;将含有枯草芽孢杆菌、白念珠菌、黑曲霉、枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢的混合溶液转移至TSB 培养基225 ml 中,于23 ℃培养不少于5 d,作为试验组;同时,以灭菌纯化水作为空白对照组、以pH 10.0缓冲液作为试剂对照组,同法操作。
表4 pH 10.0 缓冲液溶解注射用微球代表性品种后无菌检查方法的适用性试验结果1)
结果如表4 所示,上述2个品种的注射用微球在45 ℃、pH 10.0 碱性环境下振荡孵育,辅料可加速降解以制备供试液进行无菌检查,由于此条件下药典规定的试验菌(营养体) 生长状态不稳定,且由于生孢梭菌为严格厌氧菌,在有氧环境下无法生长(见表4 空白对照组),因此可选择枯草芽孢杆菌芽孢和短小芽孢杆菌芽孢作为试验菌,进行方法适用性试验。
3、讨论
本研究在调研注射用微球生产工艺和产品注册申报资料的基础上,选择注射用醋酸亮丙瑞林微球等代表性品种,建立了2 种无菌检查方法:一是采用DMSO 溶解微球;二是采用pH 10.0 水溶性缓冲液加速辅料降解后溶解微球。制备的无菌检查供试液依照ChP 2020 通则1101,以枯草芽孢杆菌芽孢或短小芽孢杆菌芽孢作为试验菌,采用直接接种法进行无菌检查。
基于注射用微球的特点,该制剂无菌检查主要关注以下技术环节。
①在充分了解产品生产工艺、质量标准、过程控制中微生物污染关键环节的基础上,经方法验证及相关方法适用性试验确认后,建立无菌检查方法;例如,目前绝大部分注射用微球产品采用无菌生产工艺,微球内部不易有微生物营养体存活,可采用枯草芽孢杆菌芽孢或短小芽孢杆菌芽孢作为试验菌,或根据产品生产工艺中潜在污染物的风险评估,采用其他耐受性芽孢作为试验菌进行方法验证。
②对于有必要进行内部无菌检查的品种,应选择适宜的溶剂制备供试液,模拟潜在微生物污染物存在条件下的微球溶解过程,并对溶剂是否影响试验菌生长进行考察;例如,采用DMSO溶解,或采用碱性(pH 10.0) 缓冲体系使辅料加速降解制备供试液,均是对特殊无菌制剂建立无菌检查方法的有益尝试。
③由于PLGA 单体比例不同,形成制剂时聚合物的降解时间、药物黏度、药物释放行为等均不相同[10—11],故不同品种在建立无菌检查方法时,需要分别对具体试验参数进行验证或确认。此外,微球骨架降解行为的目测判断并不充分,如某些品种,在一定的加速释放介质中发生“溶蚀”现象,虽然辅料载体未完全溶解(或降解),但微球由“光滑球面”逐渐变为“蜂窝状多孔缝隙球面”,因此,必要时可借助SEM 进行形态观察、含量测定等方法,佐证无菌检查方法的开发或验证策略[12—13]。
注射用微球的生产工艺个性化,且相对常见注射剂,仍属于小众的特殊剂型。本研究选择注射用醋酸亮丙瑞林微球、注射用利培酮微球、注射用醋酸奥曲肽微球等代表性品种进行研究,基本覆盖了目前研发和上市的品种。除本研究研究的品种外,还存在特殊生产工艺下的剂型,如注射用全氟丁烷微球,该制剂为空心微球,根据该产品质量标准所述,可采用鲁尔接头通过单向加压方式破坏微球结构后制备供试液,进行外部和内部的整体无菌性检查。
根据ChP 2020 通则1101 有关规定,品种性状允许,应优先选择薄膜过滤法进行无菌检查。由于DMSO 对混合纤维素、聚偏氟乙烯等材质的无菌检查用滤膜具有腐蚀性,pH 10.0 缓冲液对滤膜完整性是否存在影响仍需评估,故本研究在建立无菌检查方法时,采用直接接种法。其他品种在进行无菌检查时,是否能够采用薄膜过滤法,仍需进行滤膜完整性影响因素考察。
参考文献
[1] YUE S, BOLUN Z, RUOWEI S, et al.PLGA-based biodegradable microspheres in drug delivery: recent advances in research and application [J].Drug Deliv, 2021, 28(1):1397-1418.
[2] 李 勋, 韦 祎, 马光辉, 等.缓释微球制剂的研究进展[J].北京化工大学学报(自然科学版), 2017, 44(6):1-11.
[3] 郭宁子, 辛中帅, 杨化新.微球制剂质量控制研究进展[J].中国新药杂志, 2015, 24(18): 2115-2121.
[4] 王似锦, 刘文杰, 高 春.注射用利培酮微球无菌检查法的建立[J].中国新药杂志, 2013, 22(11): 1341-1344.
[5] SUJI R, SEUNGYEOP P, YEON H L, et al.Biodegradable nanoparticles-loaded PLGA microcapsule for the enhanced encapsulation efficiency and controlled release of hydrophilic drug [J].Int J Mol Sci, 2021, 22(6): 2792.
[6] 张伊洁, 郭宁子, 刘万卉, 等.缓控释注射剂中丙交酯乙交酯共聚物(PLGA)分析方法的研究进展[J].中国医药工业杂志, 2019, 50(10): 1180-1187.
[7] 韩宁娟, 牛 睿, 葛维娟.在药剂学中微球制剂制备方法研究[J].生物化工, 2019, 5(2): 114-116.
[8] 张雪娟, 付 寒, 赵紫玉, 等.利培酮PLGA长效注射微球的稳定性考察[J].中国医药工业杂志, 2019, 50(10):1193-1200.
[9] 梁苑英竹, 袁 松, 郭宁子, 等.注射用醋酸奥曲肽微球体内外释放度分析[J].药物分析杂志, 2020, 40(6): 955-963.
[10] GUANGLIANG L, KATHLEEN M.Glass transition temperature of PLGA particles and the influence on drug delivery applications [J].Polymers, 2022, 14(5): 993-1011.
[11] 张伊洁, 郭宁子, 许丽晓, 等.多肽微球缓释注射剂中载体辅料丙交酯乙交酯共聚物的关键质量属性分析[J].药物分析杂志, 2020, 40(6): 988-998.
[12] SHAWN Z, DAN W, LIPING Z.Characterization of controlled release microspheres using FIB-SEM and imagebased release prediction [J].AAPS Pharm Sci Tech, 2020,21(5): 194-202.
[13] HONGFEI L, SHUANGSHUANG S, JIN C, et al.Preparation and evaluation of a novel bioactive glass/lysozyme/PLGA composite microsphere [J].Drug Dev Ind Pharm, 2015, 41(3): 458-463.
本文作者冯震、肖珊珊、陈辉、李静敏、唐黎明、杨美成,上海市食品药品检验研究院、上海丽珠制药有限公司,来源于中国医药工业杂志,仅供交流学习。