摘要
模拟技术是提高临床试验成功率的重要工具,且在临床研究各阶段中均有重要作用。本文阐述了模拟计划方案、执行、总结和评价3部分中的内容、要求和需关注的问题。在以生存分析的应用实践中,本文假设的情景为不符合等比例风险假设的时间事件数据,使用的模型为包含延滞效应的指数治愈率函数模型,模拟目的是对平均风险比、样本量、事件数等试验设计因素进行研究。模拟结果显示,非等比例风险假设对于试验假设、随访时间和分析时间均有重要影响。使用等比例风险假设下的试验设计方法会造成试验检验把握度降低、不可靠的期中分析结果等问题,而模拟技术可以更好地支持临床试验的设计和实施。最后讨论了模拟实施中情景假设的重要程度和对模拟执行的影响,并指出应考虑模拟的复杂度以提高模拟执行的可行性。
一、概述
模拟技术是药物临床研究中非常重要的工具,尤其在新颖、复杂的临床适应性设计研究中,模拟技术是提高试验成功率的关键手段。在应用类型方面,模拟技术可用于确定药物剂量、评估药物毒性、研究剂量-疗效关系、确定期中分析时间节点、样本量重新估计等。通过对试验过程的模拟,研究者可以预估试验中受试者的入组速率和随访时间,统计师可以设定临床试验的期中分析策略,申办方可以规划相应的药物临床开发战略[1]。
模拟技术作为新颖临床研究的重要组成部分,其设计和执行非常重要。近些年,各国监管机构制定的指导原则中均包含了相关要求。国家药品监督管理局药品审评中心(Center for Drug Evaluation,CDE)于2020年12月发布的《模型引导的药物研发技术指导原则》[2]中对于建模与模拟实施的过程和报告结构进行了规范。美国FDA在2019年发布的《适应性设计指导原则(Adaptive Design Clinical Trials for Drugs and Biologics Guidance for Industry)》[3]中规定了模拟报告的格式和内容。次年,美国针对复杂新颖试验设计(complex innovative trial design,CID)项目制定了《与FDA沟通交流药物和生物制品复杂创新试验设计指导原则(Interacting with the FDA on Complex Innovative Trial Designs for Drugs and Biological Products)》[4],其中对沟通交流时模拟技术内容和要求进行了细化。欧洲EMA和欧洲制药工业和协会联合会(European Federation of Pharmaceutical Industries and Associations,EFPIA)成立的模型和模拟工作组于2016年发布了《模型引导的药物发现和与研发(Model-informed Drug Discovery and Development,MID3)良好做法》白皮书[5],其中制定了模拟计划书和报告内容。
在学术层面,已有学者对模拟技术的良好操作流程(good practices)进行了总结[6],之后伴随临床研究的发展和进步,又对模拟技术进行了深入研究[7-9],进一步细化了模拟技术的实施过程,并探索了模拟技术的影响因素。
本文将从具体实施过程角度阐述模拟技术中设计和执行的重要性。之后介绍一项在非等比例假设下生存分析试验设计中模拟技术的应用实践。
二、模拟技术的实施
在临床研究中,模拟技术的复杂程度会随着其重要程度的增加而增加[10]。如果模拟技术的重要程度较低,则其设计和实施相对而言会较为简略; 若模拟技术的结果对于临床研究的设计和决策有较大意义,则其设计和实施需要较为规范和具体的形式。尽管模拟技术形式多样,但其核心理念基本相同。本节将按照以下3部分阐述: 模拟的计划、模拟的执行、模拟的总结和评价。
2.1 模拟计划方案
模拟计划方案是模拟实施的关键前提,完善的计划可保证模拟实施过程的合规性和模拟结果的可靠性。模拟分析计划不仅由统计师制定,其中的细节内容应由临床专家和实施人员共同审阅和完善。完备的模拟实施计划主要包含3个部分[10]: 首先明确模拟在临床研究中的地位和价值、模拟结果如何指导临床研究的设计和实施; 其次阐述模拟过程中的模型假设、建模方法、模拟情景、软件使用等基本内容; 最后说明模拟结果的评价和解读。
监管机构在指导原则中对模拟分析计划进行了说明。CDE发布的《模型引导的药物研发技术指导原则》中明确模型分析计划应包含背景、目的、研究概述、分析用数据、分析方法5部分。美国FDA发布的《适应性设计指导原则》中说明,若模拟对于试验实施参数具有非常重要的作用,则其设计需要被详细描述,包括模拟情景参数的设置、模拟次数或迭代次数、所使用的软件包、随机数产生的过程等。欧洲EMA发布的白皮书中着重阐述了模型方法的内容,包括对于干扰项等重要内容的敏感性分析等,同时强调了情景假设对于模拟的重要程度。
模拟的本质是使用数学模型替代实际临床试验,观察临床试验在不同情景下的运行情况,因此模拟的关键内容之一就是情景假设(scenario assumptions)。情景假设包含模型假设和参数假设。模型假设是指针对临床研究中各因素间关系而近似表述出的数学结构,如代表药动学的一房室模型、二房室模型; 生存分析中的比例风险模型(Cox)等。参数假设是指模型中参数的假设,其可以假设为具体值或某类分布,例如: 房室模型中消除速率设定为某常数,生存分析中事件发生率服从指数分布等。参数假设的来源包括临床研究、非临床研究以及文献资料等,其证据质量需得到充分保证。模拟的情景假设应建立在临床研究基本特征的基础上,且需要考虑到模型和参数假设背后的统计学因素。
模拟结果的统计分析和评价是另一重要内容。模拟与实际临床研究的统计分析原则基本是一致的,差异主要体现在模拟产生偏倚的来源可能与实际临床研究不同,而这会在一定程度上影响统计分析结果的稳定性。因此在模拟的统计分析中需对模拟造成的偏倚进行额外考虑或进行更多的敏感性分析。模拟通常会在不同的情境下进行多次模拟,并根据模拟结果选择更适合的临床情景。例如: 模拟不同入组时间和随访时间以找到最为合适的期中分析时间点。不同情景下模拟结果的评价工具是操作特征(operating character),常用的操作特征包括Ⅰ类错误、检验效能、成功概率(probability of success,POS)等。
当不能保证参数假设的数据质量时,可尝试使用贝叶斯方法迭代更新参数分布以收敛至稳健结果。此时需要对模拟误差进行估计,并阐述迭代次数的合理性,如参数估计随着迭代如何收敛。
随着模型复杂性及模拟次数的增加,常规统计分析软件可能无法满足模拟的需要。因此,需要在模拟计划时阐述所使用软件及相应软件包的名称、版本以及在此基础上的程序代码内容。涉及随机数的产生还需说明随机数种子数,以便后期审查时重现随机过程。
2.2 模拟的执行
药物临床试验应当遵守药物临床试验质量管理规范,同样,模拟的执行过程也应处于严格的质量控制以保证模拟结果准确可靠,尤其应重点关注模拟的流程控制。模拟的整个执行过程应是可追溯的,并保留稽查轨迹和操作轨迹。此外,与实际临床研究数据搜集相同,模拟的执行中也应对产生的数据进行良好的保存和处理,以确保情景假设被准确实现。尤其在使用贝叶斯统计进行参数估计时,迭代过程中的数据质量对于参数是否可以收敛是非常重要的。
模拟计划经确定后,在实施过程中一般不得更改。对于任何的修改应在修订方案中写明,且一般应经再次审核。为进一步确保模拟执行的质量,建议由未参与模拟执行的专业人员对过程进行独立审核,包括从最初情景假设呈现到最终结果生成的全流程以及程序代码的准确性。
2.3 模拟结果的总结与评价
国家药品监督管理局(NMPA)和欧洲EMA的指导原则均对模拟总结报告的内容和格式有详细要求,且两者结构较相似。总结报告应提供模拟过程和结果的详细内容并可被重现,以确保监管机构可基于此报告作出科学准确的判断。在结果部分中,应先展示最终选择的情景假设内容,包括模型假设和参数假设,以及在当前情景下的直接模拟结果和进一步的统计分析结果。若存在其他敏感性分析内容,也应在结果中展示。
在讨论和结论中,应分别从临床和统计方面对模拟结果进行解释说明,同时还需说明不同模型假设和参数假设对模拟结果的影响。模拟研究通常作为临床研究决策的支持性证据,其结果存在不确定性,因此应充分讨论模拟结果对决策的支持性作用,并综合评估相应风险。例如: 讨论最初使用原始数据的充分性和代表性、情景假设的合理性、统计分析的科学性、模拟结果的可解释性和对决策的适用性等。
三、应用实例: 非等比例假设下的生存分析设计
3.1 相关背景
生存分析被广泛应用于抗肿瘤药物临床试验评价,是作为时间-事件(time-to-event)数据的主要分析方法,如总生存期(overall survival,OS)、无进展生存期(progression free survival,PFS)、无疾病生存期(disease free survival,DFS)。
在生存分析中,通常假设时间-事件数据符合等比例假设,即试验组和对照组的风险比随时间变化是恒定的。而在免疫治疗中,由于延滞效应等因素,其时间事件数据通常不满足等比例风险假设,例如: 一项在转移性黑色素瘤中比较纳武利尤单抗与伊匹木单抗的研究表明[11],纳武利尤单抗存在3个月的延滞效应。而使用基于比例风险假设的统计学方法应用于非比例风险假设的时间事件数据时会产生不适用问题,包括统计把握度的降低、不可靠的期中分析结果、最终结果的解释困难等。
3.2 情景假设
假设一项在某癌症中比较免疫抑制剂A与安慰剂辅助治疗的随机对照研究,使用DFS作为主要疗效指标,并在α为2.5%(单侧)的情况下进行优效性检验。而由于免疫抑制剂辅助治疗中DFS数据通常不满足非等比例假设的情景,常规试验设计存在把握度较低的情景,因此使用模拟技术的目的在于寻找最优的事件数和相应样本量组合。
假设DFS数据不符合等比例假设的原因主要在于: ①延滞效应。②存在一定数量的无事件者导致两组的DFS曲线在末端变平。这导致试验组和对照组的风险比在各个时间点不同,例如: 在试验初期风险比均为1,而在试验中期风险比随时间变化呈非线性变化并固定于某值。
针对非等比例假设情况,假设以下情景: 延滞效应发生的时间为前3个月; 使用指数治愈率函数描述尾端DFS曲线变平情况,即:
其中p表示治愈率,α表示未治愈人群指数分布。假设免疫抑制剂率的治愈率为45%,安慰剂组的治愈率为30%; 假设免疫抑制剂组和安慰剂组的未治愈人群分布相同,其参数α均为0.1。其他情景假设如下: 受试者按照1∶1的比例分配至试验组和对照组; 总体Ⅰ类错误率为2.5%(单侧); 受试者入组率为每月20例。
由于假设DFS曲线为存在延滞效应的指数治愈率函数,则试验组和对照组的平均风险比(hazard ratio,HR)在理论上处于0.67(0.3/0.45)至1之间。需要注意的是,试验持续时间不可过短也不可过长: 若试验完成时间过短,则平均HR受延滞效应影响过大,无法充分体现试验组的有效性; 若试验完成时间过长,虽然其平均HR无限接近于0.67,但是其可行性较低。因此需首先探索可行的平均HR值,之后再针对不同样本量下的事件数进行模拟,以选择最优的样本量和事件数组合。模拟使用检验效能和随访时间作为操作特征对模拟结果进行评价。
模拟的实施采用R软件,并调用其中的Hmisc包随机生成生存分析时间数据[12]。每个情景下模拟次数为1000次,种子数设置为20220701。
3.3 模拟结果与结论
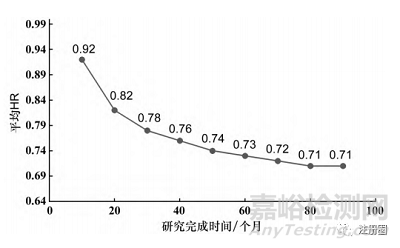
首先在充足样本量的情况下(1000例),探索研究完成时间10~90个月情况下的平均HR,模拟结果见图1。
▲图1-不同研究完成时间下的平均 HR
从图中可以看到,在研究时间仅为10个月的时候,试验组和对照组的平均HR为0.92,即严重受到3个月延滞效应的影响; 当研究时间>60个月时,试验组和对照组的平均HR下降趋势逐渐平缓至0.71。因此基于实际操作考虑,可以认为平均HR≤0.73为合理的试验假设情景。
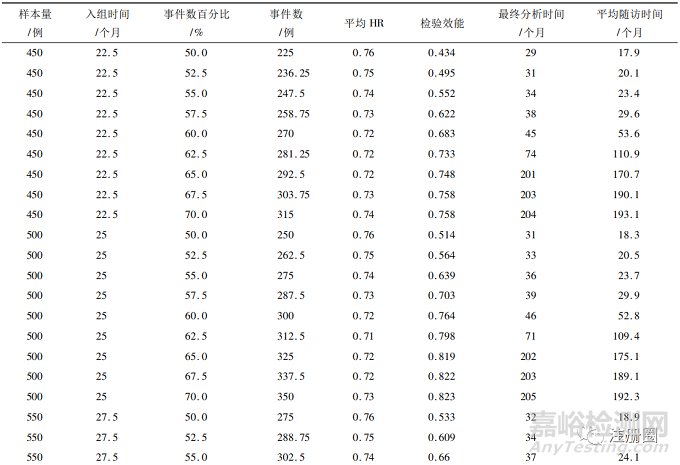
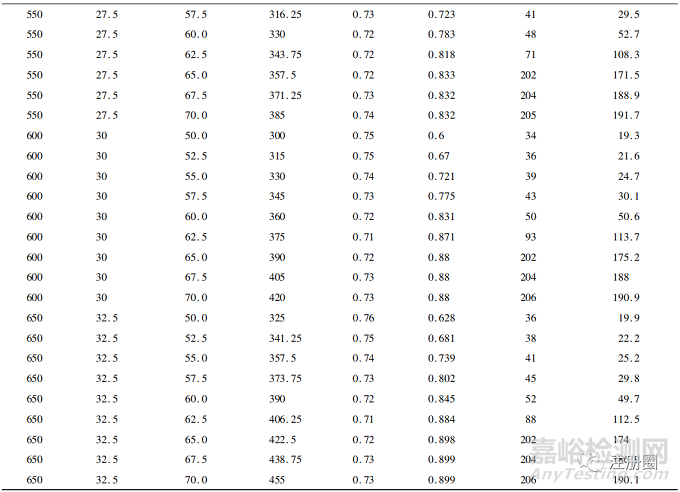
在此基础上,分别模拟450~650个样本量下50%~70%事件数的情况,结果见表1。
▲表1-不同样本量及事件数下模拟结果
清晰版本图片,详见《中国新药杂志》 2023年第32卷第14期
从表中可以看到,当事件数>60%时,最终分析时间和平均随访时间显著增加,这是因为模拟计划中假设试验组和对照组分别有45%和30%的治愈率,因此当设置的事件数>60%时,需花费更多时间随访才会发生一例事件。在样本量为550例及以下的60%事件数时,虽然其平均HR已达到0.72,但其检验效能不足80%。在600例样本量的60%事件数(情景1)时,其检验平均HR为0.72的效能达到83.1%,在650例样本量的57.5%事件数(情景2)时,其检验平均HR为0.73的效能达到80.2%。
综上所述,情景1和情景2均能满足平均HR和检验效能的要求。在决策选择最优组合时,可结合最终分析时间和平均随访时间进行综合分析。例如: 情景1比情景2少50例受试者,若受试者入组成本较高的情况下可选择情景1; 情景2虽然增加了受试者数量,但其最终分析时间和平均随访时间均要短于情景1,因此若在试验的时间成本较高情况下,可以选择情景2。
四、讨论
在当今临床研究各方面要求提高的情况下[13-16],模拟技术可以在临床研究设计时更精确判断实施过程中各方面因素的影响,为具体的试验设计和实施提供准确指导[17-18]。模拟技术可以带来显著优势。首先,基于模拟技术可以尽早结束无价值研究,或者优化研究设计以期在更短时间和更低成本中达到预设试验目的而减少临床研究成本[19]; 其次,模拟技术可以在临床试验开展前探索不同因素对试验的影响[20],在一定限度上规避临床失败原因[21],提高临床试验成功率; 最后,在非常规临床试验条件情况下,模拟技术具有解决复杂问题能力[1],如上述非等比例假设下的生存分析试验设计。
模拟技术的实施不是盲目探索所有因素的影响情况,而应基于对情景假设的充分理解并预估出可能的结果和影响因素。在上述的应用实践中,对于非等比例情况下假设了延滞效应和指数治愈率函数,在模拟实施的研究中则要考虑到这2项假设所带来的影响。首先是试验的持续时间不可过短,以避免延滞效应影响而导致的假阴性; 同时也要考虑到试验组和对照组有相应的治愈率,因此设定的事件数存在上限,以避免过长的随访时间。
模拟计划方案时应考虑模拟的复杂度,尤其是模拟内容包含较多因素时。虽然充足的模拟次数可以得到可靠的结果,但是复杂因素下的一次模拟可能需要几小时甚至几天。因此模拟计划需充分考虑模拟的可行性,并可通过预试验或预模拟尽可能地缩小情景假设中的不定因素,将模拟内容聚焦于最需了解的内容。在本文的应用实践中,仅针对样本量和事件数进行了模拟,其他情景假设为固定值或较为简单的情景,例如假设受试者入组时间为每月20例,但在实际临床研究中受试者的入组速度并不完全符合均匀分布,再例如模拟中并未假设脱落等删失情况。因此在使用模拟技术的结果进行决策时,应充分考虑到实际问题。
药物临床试验的根本目的在于评价和确定药物的临床疗效,从而综合判断药物的风险获益。模拟技术虽然在一定限度上可以提升临床试验的成功率,但仅依靠模拟技术得到的临床结果或使用模拟技术反转临床研究的无效结论是不可接受的。因此在使用模拟技术时,应根据整体药物研发路径和当前所处阶段,确定模拟技术的具体作用和产生结果的证据强度。
参考文献
[1] ABBAS I. Modeling and simulation in clinical trials[R]. SpringSim ( MSM) . 2016: 1.
[2] 国家药品监督管理局药品审评中心. 模型引导的药物研发技术指导原则[EB/OL]. ( 2020 - 12 - 31) [2021 - 12 - 23]. https: / /www. cde. org. cn /zdyz/domesticinfopage? zdyzIdCODE =e0651af6eba8cc2 f5f31efb7add1f0a0.
[3] Food and Drug Administration ( FDA) . Adaptive Design ClinicalTrials for Drugs and Biologics Guidance for Industry[EB/OL].( 2019 - 11 - 29) [2021 - 12 - 23]. https: / /www. fda. gov /regulatory-information /search-fda-guidance-documents/adaptive-design-clinical-trials-drugs-and-biologics-guidance-industry.
[4] Food and Drug Administration ( FDA) . Interacting with the FDA on Complex Innovative Trial Designs for Drugs and Biological Products[EB /OL]. ( 2021 - 01 - 22) [2021 - 12 - 23]. https: / /www. fda. gov /regulatory-information /search-fda-guidancedocuments/interacting-fda-complex-innovative-trial-designs-drugs and-biological-products.
[5] EFPIA MID WORKGROUP,MARSHALL SF,BURGHAUS R,et al. Good practices in model-informed drug discovery and development: practice,application,and documentation[J]. CPT Pharmacometrics Syst Pharmacol,2016,5( 3) : 93 - 122.
[6] GAL J,MILANO G,FERRERO JM,et al. Optimizing drug development in oncology by clinical trial simulation: why and how?[J]. Brief Bioinform,2018,19( 6) : 1203 - 1217.
[7] SMITH MK,MARSHALL A. Importance of protocols for simulation studies in clinical drug development[J]. Stat Methods Med Res,2011,20( 6) : 613 - 622.
[8] GIOVAGNOLI A,ZAGORAIOU M. Simulation of Clinical Trials: a review with emphasis on the design issues[J]. Statistica,2012,72( 1) : 63 - 80.
[9] O’KELLY M,ANISIMOV V,CAMPBELL C,et al. Proposed best practice for projects that involve modelling and simulation[J]. Pharm Stat,2017,16( 2) : 107 - 113.
[10] O’KELLY M,ANISIMOV V,CAMPBELL C,et al. Proposed best practice for projects that involve modelling and simulation[J]. Pharm Stat,2017,16( 2) : 107 - 113.
[11] WEBER J,MANDALA M,VECCHIO M,et al. Adjuvant thera py with nivolumab ( NIVO) versus ipilimumab ( IPI) after complete resection of stage III/IV melanoma: updated results from aphase III trial ( CheckMate 238) [J]. J Clin Oncol,2018,36( Suppl 15) : S9502.
[12] HARRELL JR FE,HARRELL JR MFE. Package‘hmisc’[J].CRAN,2019: 235 - 236.
[13] HOWIE LJ,PEPPERCORN JM. The ethics of clinical trials for cancer therapy[J]. N C Med J,2014,75( 4) : 270 - 273.
[14] SHAMY MCF,STAHNISCH FW,HILL MD. Fallibility: a new perspective on the ethics of clinical trial enrollment[J]. Int J Stroke,2015,10( 1) : 2 - 6.
[15] ROSENBLATT M. The large pharmaceutical company perspective[J]. N Engl J Med,2017,376( 1) : 52 - 60.
[16] TANG C,SHERMAN SI,PRICE M,et al. Clinical trial charac teristics and barriers to participant accrual: the MD Anderson cancer center experience over 30 years,a historical foundation fortrial improvement[J]. Clin Cancer Res,2017,23( 6) : 1414 -1421.
[17] 李若冰,言方荣,王骏. 贝叶斯统计在儿童用药有效性外推中的应用[J]. 中国医药工业杂志,2022,53 ( 11) : 1606 -1611.
[18] 李若冰,李健,王骏. 模拟技术应用于临床研究的监管现状及阿达木单抗案例分析[J]. 中国新药杂志,2023,32( 2) :198 - 204.
[19] GIRARD P,CUCHERAT M,GUEZ D,et al. Clinical trial simulation in drug development[J]. Therapie,2004,59 ( 3) : 287 -295,297 - 304.
[20] VEYRAT-FOLLET C,BRUNO R,OLIVARES R,et al. Clinical trial simulation of docetaxel in patients with cancer as a tool for dosage optimization[J]. Clin Pharmacol Ther,2000,68( 6) : 677 - 687.
[21] GIRARD P. Clinical trial simulation: a tool for understanding study failures and preventing them[J]. Basic Clin Pharmacol Toxicol,2005,96( 3) : 228 - 234.