依那普利是临床上常用的血管紧张素转换酶抑制剂,用于各种原发性高血压、肾性高血压、充血性心力衰竭。原料药主要以马来酸依那普利的形式存在,原料存在的主要杂质为依那普利拉(杂质Ⅰ)和依那普利双酮(杂质Ⅱ),其中主要是杂质Ⅰ。
本文拟以欧洲药典的方法为基础,对其进行色谱条件优化,进行杂质定量测定方法的验证,以期为杂质Ⅰ提供可靠的定量检测方法,同时为该类方法的验证提供更加科学全面的验证参数。
1、 仪器和试药
1.1 仪器
XP205DR电子天平;2台岛津LC20A液相色谱仪;Mili-QIntegrals超纯水系统;酸度计;KQ-300DV型数控超声波清洗仪。
1.2 试剂和试药
对照品:马来酸依那普利(批号100705-201604,含量99.9%)、依那普利拉(杂质Ⅰ)(批号2016-784,含量88.1%)。
样品:马来酸依那普利原料(批号5113-6-015)。
试剂:磷酸(分析纯,批号20171026);磷酸二氢钾(分析纯,批号20161208;乙腈(色谱级,批号171011);甲醇(色谱级,批号163201)。
2、 流动相及其他溶液的配制
2.1 流动相及溶剂
缓冲液:称取磷酸二氢钾5.4g,加水制成1000mL,用磷酸调节pH至3.0;溶剂1:缓冲液-乙腈-甲醇(1∶2∶2);溶剂2:溶剂1-缓冲液(8∶92);流动相A:溶剂1-缓冲液(1∶9);流动相B:乙腈。
2.2 供试品储备液
精密称取马来酸依那普利原料适量,置50mL量瓶中,加甲醇10mL,加溶剂2稀释至刻度,制成20mg·mL-1的供试品储备液。
2.3 对照品溶液
精密称取依那普利拉对照品适量,置20mL量瓶中,加甲醇2mL,加溶剂2稀释至刻度,制成1mg·mL-1的对照品储备液。分别精密量取对照品储备液1、2和3mL至100mL量瓶中,加溶剂2稀释至刻度,分别制成质量浓度为10、20和30μg·mL-1的对照品溶液。
2.4 混合对照品溶液
精密称取马来酸依那普利对照品和依那普利拉对照品至10mL量瓶中,加甲醇2mL,加溶剂2稀释至刻度。精密量取1mL至100mL量瓶中,加溶剂2稀释至刻度,制成20μg·mL-1的混合对照品溶液。
3、 色谱条件及测定方法
3.1 色谱条件
采用ShiseidoC18色谱柱(4.6mm×250mm,5μm),柱温65℃,进样器温度4℃,进样量20μL,流动相同“2.1”项,梯度洗脱,洗脱顺序见表1,流速1mL·min-1,检测波长210nm。
3.2 测定法
精密称取马来酸依那普利适量,加甲醇2mL,加溶剂2稀释至刻度,制成每1mL中约含2mg的溶液,作为供试品溶液,精密量取20μL,注入液相色谱仪,记录色谱图;另取依那普利拉对照品适量,精密称定,加甲醇6mL,加溶剂2制成每1mL约含6μg·mL-1的对照品溶液,同法测定,按外标法计算含量,依那普利拉的含量不大于0.3%。
4、 统计分析软件
本文所用统计软件包括JMP和理化方法验证统计软件(PCMV)。
5、 方法验证及结果
5.1 系统适用性试验
取“2.4”项下的混合对照品溶液,按照“3.1”的色谱条件进样,获得的色谱图见图1,出峰顺序为马来酸、依那普利拉和依那普利。依那普利峰的拖尾因子为1.0<2.0,马来酸与依那普利拉分离度19.0>1.5,依那普利拉与依那普利分离度为30.9>4.0,依那普利拉理论塔板数不低于5000。
5.2 专属性
取“2.4”项下的混合对照品溶液,按照“3.1”的色谱条件,进样,获得的色谱图如图1所示,马来酸与依那普利拉分离度19.0>1.5,依那普利拉与依那普利分离度为30.9>4.0,方法的专属性良好。
5.3 线性、检测下限与定量下限和范围
5.3.1 实验设计
在浓度范围为其质量限度标准(0.3%)的8.0%(预实验确定的LOD)~200.0%内设置9个浓度点,0.5、1.0、2.0、3.0、4.0、6.0、8.0、10.0、12.0μg·mL-1,每个浓度独立制备3份。
5.3.2 操作步骤
精密量取“2.3”项下10μg·mL-1的对照品溶液0.5、1和2mL,20μg·mL-1的对照品溶液2、4、5mL,30μg·mL-1的对照品溶液1、2和4mL,分别置10mL量瓶中,加溶剂2稀释至刻度,制成约0.5、1.0、2.0、3.0、4.0、6.0、8.0、10.0、12.0μg·mL-1的对照品溶液,每个浓度独立制备3份,进样测定,试验结果见表2。
5.3.3 线性数据分析
线性方程:以浓度X为横坐标,峰面积Y为纵坐标,得浓度-峰面积的最小二乘回归图,见图2。线性方程:Y=139.86+38982.17X r=0.9998截距估计值:截距为-139.86,截距的标准误为867.04,t比为-0.16,概率>|t|值为0.87>0.5,统计上显示截距与0没有显著性差异,由此确定依那普利拉杂质可通过外标法确定其含量。回归线的标准误为2695.87。
5.3.4 检测下限与定量下限
基于“5.3.3”项的回归方程,采用线性 95% 的预测区间确定分析方法的检测下限与定量下限分为:
5.3.5 线性范围
该分析方法在0.5~12.0μg·mL-1的浓度范围内均满足线性要求。
5.4 准确度和精密度
5.4.1 实验设计
(1)对影响此方法的因素进行风险分析,确定本方法的主要影响因素:仪器、分析人员以及每个分析人员的独立操作次数(时间);
(2)通过预实验,确定偏倚约为10%,变异约为5%,将α设为5%,效能设为90%时,通过JMP软件求得所需的样本量,即实验次数不少于5次;
(3)以上述2个条件为基础,将3个主要因素均设置2个水平,采用3因素全析因
设计,确定8个实验条件(见表4),满足参数计算的样本量需求;
(4)浓度范围选择:以加标浓度为其质量限度标准(0.3%)的8.0%(预实验确定的LOD)~150.0%为参考,设置7个浓度点:0.5、1、2、4、6、8、9μg·mL-1。
5.4.2 操作步骤
精密量取“2.3”项下10μg·mL-1的对照品溶液0.5、1、2mL,20μg·mL-1的对照品溶液2、4mL,30μg·mL-1的对照品溶液2、3mL,分别置10mL量瓶中,分别加“2.2”项下的供试品储备液1mL,加溶剂2稀释至刻度,制成加样浓度约为0.5、1、2、4、6、8、9μg·mL-1的溶液,每个浓度独立制备3份。按照“3.1”项下的色谱条件进样。结果汇总于表4。
5.4.3 结果分析
5.4.3.1 精密度
通过对每个浓度进行方差成分分析,可获得该分析方法在不同浓度下的重复性,中间精密度及其精密度上限值。设α=0.05,精密度结果表见表5。
5.4.3.2 准确度
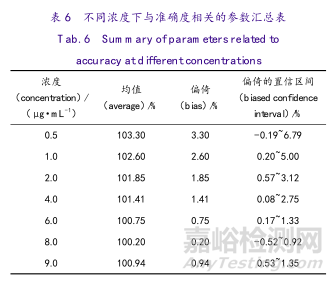
通过对每个浓度下真实值(100%)与回收率结果的差值获得方法在该浓度下的偏倚值,以及获得其置信区间。设α=0.05时的结果分析表见表6。
5.4.3.3 方法能力的综合评价
对于准确度与精密度的联合判定,引入新的统计评价参数,方法变异、方法能力指数(MCI)、方法误判率、方法分级、预测区间和容忍区间。假定杂质的质量标准在90.0%~110.0%时能满足产品检测的需求,则分析结果见表7。
图3和图4为在不同浓度下的预测区间或容忍区间与质量标准的关系图。从表7、图3和图4可以看出,此分析方法在2.0~9.0μg·mL-1时,方法的准确度与精密度能满足产品检测需求。
5.5 方法适用范围
在准确度、精密度、LOQ和线性满足前提下的方法适用浓度范围为2.0~9.0μg·mL-1。
5.6 溶液的稳定性试验
一次进样需运行45min,为保证在方法验证期间,所配制溶液的稳定性,考察了供试品溶液在60h内的稳定性。
精密量取“2.1”供试品储备液1mL至10mL量瓶中,加溶剂2稀释至刻度,在0、5、10、20、30、40、50、60h按照“3”项色谱条件进样,获得依那普利和依那普利拉的峰面积。从表中可以看出,随着所配制溶液放置时间的延长,依那普利拉逐渐增多,依那普利减少。在40h内,依那普利拉峰面积的RSD=0.72%,溶液维持稳定。
6、 讨论和结论
6.1 马来酸依那普利原料药中依那普利拉杂质定量测定的检测方法
2015年版《中国药典》中对马来酸依那普利原料药中依那普利拉杂质进行的是限度检查,该方法的系统适用性试验在主成分和杂质组分均为20.0μg·mL-1时满足药典要求。但在进行实际产品检测时,由于试验时按2.0mg·mL-1浓度进样(高于系统适用性浓度100倍),导致马来酸峰与依那普利拉峰出现一定程度重合(图5),使含量计算存在被低估的风险。
为使马来酸峰与依那普利拉峰能完全分开,保证能够准确定量依那普利拉峰,作者以欧洲药典中原料药依那普利拉的杂质进行限度测定方法的色谱条件为基础进行优化,综合考虑峰形、马来酸峰与依那普利峰的分离度以及分析时间等因素,最终确定的色谱条件。
该方法的缺点是无法检测到马来酸依那普利中的杂质依那普利双酮,但本实验主要用于对杂质类定量分析方法性能参数的评估探讨,故不影响此目的的阐述。
6.2 LOD和LOQ的确定
目前ICHQ2(R1)指南和专著给出的确定LOD与LOQ的方法可以归为4类:
1)可视化的评估;
2)基于空白分布;
3)基于线性;
4)基于分析方法的精密度。
国内最常应用的是基于空白分布中信噪比法来确定LOD与LOQ。本文所计算LOD的方法是基于USP<1210>提出的线性95%的预测区间来确定,此方法不仅考虑了Ⅰ类错误与Ⅱ类错误,同时将实验测定的变异性(不确定度)考虑在内。因此更加可靠和科学。
按照<1210>的阐述,该方法所得LOD可作为最小LOQ使用,但为保证LOQ更加可靠,本实验将LOQ用10替代计算公式中的(t1-α;n-2+t1-α;n-2)。
6.3 准确度和精密度的获取和描述
目前国内有关方法验证的文章中通常对方法准确度的评价仅提供偏倚及其置信区间,无法从整体体现方法的变异。系统精密度和重复性的验证,通过对某一对照品溶液连续进样6针来确定仪器精密度,将同一供试品溶液平行制备6份来确定方法的重复性。该方法节省时间成本,但是不能够正确表明方法在不同浓度下的变异性。
本文中有8个实验条件,每个实验条件下对每个浓度独立制备3份溶液,通过方差成分分析可以同时确定中间精密度与重复性。同理对于重复性和仪器精密度也可以按照本文的方法确定。本文的试验设计充分考虑了不同浓度下该方法的变异,从整体把握方法的适用性及其适用范围。
为了更充分表达所获准确度和精密度,本文所采用的设计,还能有效地计算出准确度置信区间和精密度置信上限,从而为技术人员和监管者更好地把握所得参数的可靠性提供依据。
6.4 方法能力综合评价参数及其比单独使用准确度
和精密度评价的优势目前ICH指南、《中国药典》和欧洲官方实验室都使用准确度和精密度单独的标准进行方法验证的判定。这里的限度标准多通过经验确定出,主观性较大,且没有与所检测产品的质量标准直接联系。
本次新增方法变异、预测区间和容忍区间、方法能力指数等综合评价方法满足其预期用途的参数(有关其详细含义请参见参见USP<1210>和本专栏《定量理化分析类方法满足预期用途的判断标准探讨》),是对准确度和精密度进行合成后衍生出的新参数,这些参数是对方法整体评价更全面的把握。
此外,将各浓度下计算的容忍区间和预测区间按浓度-质量标准限度进行构图(见表7和图3和图4),可更清晰和直观地了解方法在不同浓度下的判断,具有科学、直观的优势。
本方法对依那普利拉杂质的检测,在LOQ0.8~2.0μg·mL-1的范围,只可确保标示量的±20%的变异内检测准确,在2.0~9.0μg·mL-1浓度范围,其容忍区间均包含于±10%以内,且对产品质量限度标准为6.0μg·mL-1浓度周围,该方法的变异容忍区小于±5%,方法能力指数达到1.02,因方法变异产生的误判率为0.2%;故认为是一个非常可靠的定量检测方法。
本次方法验证所获预测区间和容忍区间不仅可用于新方法进行方法验证,也为后续方法的转移和验证判定提供依据,即方法转移或确认时,进行一次实验所获得的结果应分布于预测区间内,所有试验结果的90%应处于容忍区间内。
6.5 结论
该方法满足对马来酸依那普利原料药中依那普利拉杂质进行定量检测的要求。在专属性方面,依那普利拉峰与马来酸峰和依那普利峰间的分离度分别为19.0和30.9。在线性、LOD和范围方面:在0.5~1.2μg·mL-1的浓度范围内呈线性相关,线性方程为Y=139.86+38982.17X,r=0.9998;残差标准误为2693.44,且残差呈随机分布,截距与零无显著差异。
由此确定杂质依那普利拉可通过外标法确定其含量;基于线性95%的预测区间确定的LOQ为0.76μg·mL-1。在综合能力评价方面:2.0~9.0μg·mL-1的浓度范围内,方法的预测区间与容忍区间均分布于限度标准的90.0%~110%内,在产品质量标准限度6.0μg·mL-1周围,方法可达到Ⅲ级方法,因方法产生的误判率仅为0.2%。溶液稳定性:在40h内溶液维持稳定。
文章来源:《药物分析杂志》2019年第39卷第2期
原标题:《杂质类定量分析方法验证中的统计学评价》
作者:朱容蝶 ,耿颖 ,谭德讲,杨化新,何兰,刘万卉(中国食品药品检定研究院)