问题1:稳定性研究的“代表性批次”如何定义和选择?
答:所谓“代表性批次”,本质要求是它的研究对象和研究目的是相匹配的。
当我们研究这个原料药的降解途径和降解杂质时,原则只需要一批经结构确证没问题,且纯度和含量较高的样品即可;而当我们研究这个原料药的效期和包装时,则至少需要一批与商业化生产相同工艺和规模的样品来进行考察,因为不同工艺和规模的样品中所包含的杂质种类和大小是不同的。另外有时候原料药的稳定性不好,并不一定是原料药本身的问题,而是其中杂质的影响。
举个例子:当我们使用丙酮作为结晶溶剂时,若原料药本身又是一个盐酸盐或硫酸盐等,在酸性环境下丙酮会发生聚合等一系列反应,从而导致出现外观颜色的变化,最终导致稳定性失败。
所以稳定性研究并不是一味的需要大家使用正式验证批的样品才行。包括从稳定性承诺的内容中我们也知道官方是允许使用其他批次来代替的。但对于原料药来说,至少应该是中试规模的样品。
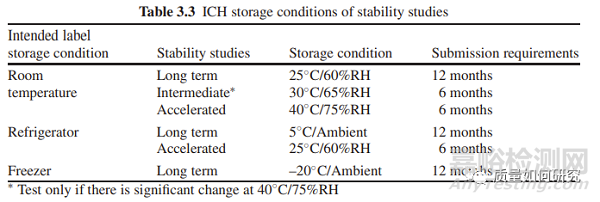
问题2:对于“拟冷冻储存”的,需不需要做加速试验?
答:对于“拟冷冻储存(即-20℃以下)”的,法规上不需要加速试验数据。但加速试验的目的不仅仅是为了辅助长期试验数据进行效期外推,还有一个重要的作用是为短期偏离提供评估数据。尤其是针对一些产品状态可能发生变化的。因此,对于拟冷冻储存的药品,仍建议大家进行适当的加速试验,通常至少考察7天以上。
问题3:稳定性研究中所用的样品,用完后如何处理?
答:对于稳定性研究用的样品,通常三批中会有一批用的比较多,比如用来进行方法学验证和影响因素考察,有时候甚至会用完了,建议大家不要销毁,比如留下外包装,至少证明它存在过。因为后面现场核查时可能会看。
问题4:影响因素中高温和高湿的条件如何选择?
答:对于高温和高湿的条件,药典和指导原则中的规定是先考察高温60℃(或高湿90%RH),若在该条件下含量明显下降(或吸湿增重5%以上),则再考察40℃(或高湿75%RH)。
首先,若大家没有前期的研发稳定性数据,则建议大家同时进行两个条件的考察,即60℃和40℃(或90%RH和75%RH)同时做。其次,若在两个条件下均出现明显变化(或吸湿增重超5%),则需要大家对原料药的包装和储存条件进行相应的考虑。
另外对于高湿试验,建议在稳定性箱中做,因为高湿溶液对于湿度的控制并不理想。
问题5:稳定性研究中,质量标准如何选择?
答:对于稳定性研究中质量标准的选择,原则上是不低于注册申报标准及企业内控标准。大家需要注意的是,此处“不低于”的含义是指限度指标的不低于,而非检查项目的不低于。
举个例子:某原料药中的氧化杂质X有明显的增长趋势,原料药的注册标准中规定该杂质不得过0.10%,企业内控标准中规定不得过0.05%。此时,稳定性考察中有关物质项下,氧化杂质X的限度应设置为0.05%或更低。
而对于检查项目的选择,稳定性研究的重点是降解产物。因此,对于那些明确不会增长或变化的检查项目则应合理删减,如残渣、重金属等,实际上变为了最低要求。
问题6:稳定性研究中,产品的效期如何推算?
答:稳定性研究的主要目的之一便是确认产品的有效期或复验期。具体规则指导原则和前面文章中有,这里简单在总结一下:
稳定性考察的3批样品,若差异小,则取平均值;若差异大,则取最短的;如没什么变化,则不做统计分析。(对于统计方法,需要保持一致,即使用同一种方法完成所有评价)
拟室温储存的,先看加速,再看中间(若有),最后看长期。
拟冷藏储存的,加速条件3~6月内发生变化,不宜外推;3月内发生变化,则需要对短期偏离进行加速评估。
拟冷冻储存的,长期数据来定效期,同时需要在5℃或25℃下对短期偏离进行加速评估。
问题7:稳定性考察的气候带如何选择?
答:原则是取决于产品上市市场情况。即主要根据产品的销售市场来确定。
问题8:微生物或无菌如何考察?
答:对于微生物或无菌的考察,若质量标准中有明确规定,则建议至少在0天和长期(每年)及加速6月的时候进行考察,以确认产品在整个效期考察期间的微生物情况。至于检测频率,可以更多点,只要与效期挂钩。
问题9:稳定性研究至少需多久之后才能申报?
答:根据规定,对于已有国家标准的品种,可以根据自身研究结果并参考已上市同品种的稳定性研究结果确定效期,审评中通常要求申报单位提供6个月的加速试验和至少6个月的长期试验数据。如果提供的上述资料显示样品比较稳定,则有效期通常可参照已上市同品种的有效期制定,但一般不超过2年。
对于新药,由于对其稳定性知之甚少,因此新药的有效期通常要根据长期试验的时间和结果确定,一般要求提供6个月的加速试验和至少12个月的长期试验数据。