摘要
抗体偶联药物(antibody drug conjugate,ADC)通常由抗体通过链接体与毒素小分子偶联而成,同时具备抗体的高靶向性和小分子药物的高活性,使之作为一种新兴的靶向治疗手段,在肿瘤治疗领域展现出了优秀的疗效和潜力,成为药物研发领域的新热点。目前全球已有14款ADC药物获批上市,处于临床研究阶段的ADC候选药物分子超过140个。为了进一步提高ADC药物的安全性和有效性,近年来涌现出了各种新颖的技术。本文对ADC药物分子的关键元素,包括抗体、链接体、毒素小分子以及偶联技术等方面的最新研究进展进行总结,并讨论其优缺点。期望这些讨论能够帮助增加对ADC药物研究和开发更加系统的理解,为研发出更加高效和安全的ADC药物带来一些思考。
正文
抗体偶联药物(antibody drug conjugate,ADC)作为一类新兴的大分子靶向药物,目前主要用于治疗各种肿瘤。ADC药物由靶向肿瘤细胞过度表达的表面受体的抗体、高活性的细胞毒素小分子和链接体3个部分组成。其中,抗体部分负责将ADC药物分子精准运送至靶细胞表面,链接体负责在靶细胞内或表面释放毒素小分子,而高活性的细胞毒素小分子则高效地杀伤肿瘤细胞,因此ADC也被通俗地称为“生物导弹”或者“魔法子弹”。
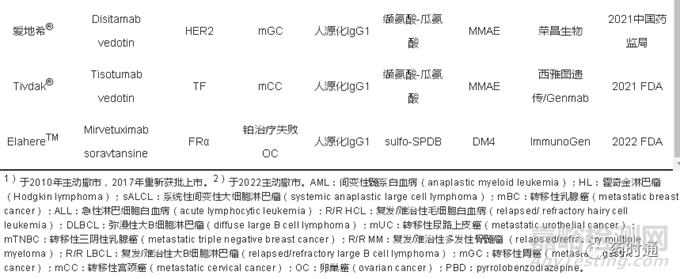
魔法子弹这一概念是由诺贝尔奖获得者德国科学家Paul Ehrlich在20世纪初提出的,特指一类能靶向结合目标病灶进而治愈疾病的一类分子。而将这一概念实物化为抗体加上毒素小分子这一药物分子形式的时间则是在1957年,Mathé等将化疗药物甲氨蝶呤通过重氮偶合反应偶联至抗L1210白血病细胞的免疫球蛋白上,该偶联物展现了针对靶细胞L1210的细胞增殖抑制作用,而偶联至常规β球蛋白的偶联物则无作用。又经过十多年的发展,在1970年代中期,有多个利用动物源免疫球蛋白制备的ADC在临床试验中展现出了确定的疗效。到1980年代,单克隆抗体开发技术和重组蛋白生产技术的进步、多种肿瘤标志物的确证,以及对靶点抗原-抗体介导的细胞内吞机制的阐明,同时在靶点、抗体以及链接体等多个方面给ADC药物的技术发展带来了巨大推动力,进而在1990年代吸引了药物研发和生物科技公司加大投入ADC药物的开发,并于2000年迎来第一个ADC药物Mylotarg®的获批上市。Mylotarg®是CD33靶向的ADC用于治疗急性髓系白血病(acute myeloid leukemia,AML)。又过去了十多年后,用于治疗霍奇金淋巴瘤(Hodgkin lymphoma,HL)和间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL)的CD30靶向的Adcetris®在2011年获批上市,用于治疗晚期转移性乳腺癌的HER2靶向的Kadcyla®于2013年获批上市。这两款ADC药物在晚期肿瘤患者中的显著疗效和良好的安全性,使得ADC药物正式作为一类新型的治疗手段得到肿瘤患者、肿瘤医疗界和生物制药工业界的广泛认可,迅速成为了全球生物医药学术界以及工业界的热点领域之一,更值得一提的是日本第一三共公司开发的HER2靶向的ADC药物Enhertu®在2019年首次获批上市,其具有划时代的意义,不仅针对HER2高表达的肿瘤有显著疗效,而且在HER2低表达晚期乳腺癌患者中也表现了明显疗效。截至2023年3月全球已有14款ADC药物获批上市(表1),其中9款是在近4年内获批的,2022年ADC药物全球销售额达到67.8亿美元,处于临床研究阶段的ADC药物分子则超过140个,而且呈现逐年快速增长的趋势有望给肿瘤患者带来更有效的治疗药物。
这些ADC药物的成功获批上市,一方面证明了此类新型药物对于肿瘤治疗的显著疗效和潜力,另一方面这些药物在临床试验和获批上市后大量病人使用过程中也逐步揭示了现有ADC药物的局限性,例如脱靶毒性、复杂的药代动力学特性、靶标组织富集度不足、耐药性、实体瘤穿透力不够等。为了进一步提高ADC药物的疗效,降低其毒副作用,各种新颖的技术和解决方案层出不穷。本文将从ADC的关键组分,包括抗体、链接体、毒素小分子以及偶联技术等方面概述其新近的研究进展,讨论目前ADC药物开发所面临的挑战和发展机遇。
经典的ADC药物由单克隆抗体、链接体和毒素小分子3部分组成(图1a)。每个组成部分发挥着不同的功能:抗体部分负责将ADC分子选择性地递送至肿瘤细胞表面,同时通过靶点介导的内吞作用进入到细胞内;链接体则负责在肿瘤细胞内或者表面高效释放毒素小分子,而在血液循环系统中保持稳定;毒素小分子则负责高效地杀伤肿瘤细胞,有些小分子还同时具备细胞穿膜渗透性,可以通过扩散效应作用于靶细胞周围的肿瘤细胞,起到“旁杀效应(bystander effect)”(图1b)。因此,ADC药物每个组成部分的特性都对其发挥靶向治疗作用至关重要,同时各个组分的选择也都可能会影响到ADC的安全性和有效性。
1、抗体
作为ADC药物的主体框架结构和导航系统,理想的抗体组分需具备较低的免疫原性、特异性的靶点结合和高亲和力、较长的半衰期、良好的血液循环系统稳定性以及能介导高效的内吞效应等特性。抗体是ADC分子的主要组成部分,占其分子质量的80%以上,ADC的潜在免疫原性主要来源于抗体。早期的ADC药物,由于采用鼠源的抗体,通常会在病人体内引发较强的免疫反应,产生抗药抗体,进而导致疗效降低和其他副反应。后来,为了克服免疫原性这一缺点,在单克隆抗体发现技术和基因工程技术进步的加持下,改进和优化出了人鼠嵌合抗体。人鼠嵌合抗体是由鼠源的抗原结合区(Fab)与人源的恒定区(Fc)嵌合而成,此种组合大大降低了其免疫原性,采用嵌合抗体的ADC也在临床上展现了良好的疗效和耐受性。第二款获批上市的靶向CD30的ADC药物Adcetris®就是采用人鼠嵌合的抗体,在治疗 霍奇金淋巴瘤(Hodgkin lymphoma)和ALCL上展现了显著的疗效。然而,也有研究显示在使用嵌合抗体治疗的病人体内,所产生的抗药抗体中的绝大部分是针对其鼠源的Fab区域,这也会影响其疗效。因此,为了进一步控制和降低免疫原性的风险,人源化抗体应运而生,它仅保留鼠源的抗原抗体结合域(complementarity determining region,CDR),Fc片段及其他框架结构则采用人源的,这样也进一步降低了抗体的免疫原性,目前已获得批准上市的14款ADC药物中,有12款采用人源化抗体。最近的一些研究则更进一步,采用全人源抗体替代人源化的抗体开发ADC药物,以期会更进一步降低其免疫原性的风险,目前这一类ADC还处于研究阶段。
除免疫原性外,抗体的高选择性和特异性也对ADC药物的安全性和有效性至关重要,它使得ADC药物能通过结合靶标而富集在肿瘤组织中,而与正常组织不结合或者较低结合。抗体的选择性与特异性,与靶点的组织分布息息相关。第一,为了降低靶点相关的毒副作用,ADC药物的理想靶点最好是仅在肿瘤细胞表达,而在正常细胞中不表达或者低表达;第二,理想的ADC靶标应为细胞表面抗原,而不是胞内抗原,使之能被循环系统中的ADC药物接触、识别并结合;第三,此靶点抗原最好不是分泌型抗原,因为分泌至循环系统中的抗原会与ADC药物结合后消耗药物而导致其可能无法以高浓度富集于肿瘤组织,影响其安全性和有效性;第四,肿瘤靶点在结合抗体或者ADC后能高效地介导内吞作用,目前主流的ADC药物发挥作用时均需要内吞至细胞内并转运至溶酶体中释放毒素小分子,才能发挥肿瘤细胞杀伤作用,如人表皮生长因子受体2(human epidermal growth factor receptor 2,HER2)就是一个理想的靶点,其在某些肿瘤细胞上的表达约为正常组织的100倍,同时它是细胞表面非分泌型受体,能高效地介导ADC药物诸如Kadcyla®、Enhertu®以及爱地希®等HER2靶向ADC药物的内吞。
ADC药物的药效与抗体-靶点抗原的亲和力密切相关,目前大部分的ADC与靶点的亲和力在0.1~1.0 nmol/L的范围内,而目前业界对于抗体最优的亲和力并没有定论。一种假设认为过高的亲和力会导致产生“结合位点屏障”,是指在应用ADC治疗实体瘤时,对靶点结合的高亲和力会导致ADC被限制在肿瘤组织表面而无法穿透到肿瘤组织内部,从而降低其治疗效果。而最近Tsumura和同事对同靶点而亲和力不同的3种ADC分子的药效和穿透效果进行了研究,结果显示高亲和力的ADC会穿透且分布在整个肿瘤组织,而低亲和力的ADC反而只能富集在肿瘤组织表面接近血管的部位,而且这种作用在体积大的肿瘤里才能表现出来。因此,在设计ADC药物时,选择靶点亲和力适当的抗体对于其内吞和药效非常关键。另外,在选择ADC药物的抗体时,针对同一靶点不同表位的抗体,其内吞效果也不尽相同。
近些年,除了利用单克隆抗体作为载具的ADC外,也有很多研究尝试使用其他形式的抗体,例如双抗、抗体片段或纳米抗体等来开发ADC,以期进一步提高ADC药物的安全性和有效性。
双抗是一种在单抗的基础上设计出的能同时识别和结合两种不同抗原或同一抗原两个不同表位的一类抗体分子。这些分子有些与传统的IgG类似,由两个Fab片段与Fc片段通过铰链区结合在一起,两个Fab片段分别识别不同的抗原或表位。另一些分子则设计得更加复杂,通过各种手段在IgG的特定位置附加上第二个抗原结合域。目前被应用于ADC药物开发的主要是与传统IgG分子结构相似的这一类双抗。目前的研究数据表明,有的双抗ADC能提高肿瘤细胞靶向性,增强靶点介导的内吞作用,从而提高药效,降低对正常组织的毒性。如Sellman等运用靶向EGFR和cMET的双抗开发出一种ADC,相比与EGFR单抗ADC或者cMET单抗ADC在同时表达两种抗原的肿瘤细胞上有更好的选择性,而且他们认为通过进一步地调整双抗两个结合臂与EGFR和cMET靶点的亲和力,能设计出靶点选择性更高的ADC,从而拓宽其治疗窗。Andreev和同事则设计了一种靶向HER2和PRLR(prolactin receptor)的双抗ADC,与针对这两个靶点的单抗ADC相比,它具备更好的乳腺癌细胞杀伤效果。同时,他们的研究还阐明了其中的作用原理:双抗ADC由于双靶点结合介导的内吞作用更快更强,因此能递送更多的ADC分子进入胞内,释放更多的毒素小分子从而更有效地杀伤肿瘤细胞。
ADC药物目前面临的挑战之一是分子质量过大,难于穿越各种生物屏障到达并穿透实体瘤肿瘤组织,影响其治疗实体瘤的效果。为了提高ADC药物治疗实体瘤的效果,研究者们希望通过缩小抗体框架结构来降低ADC的总体分子大小,增强其肿瘤穿透力。作为ADC药物的框架结构,这些缩小版的抗体首先必须保留其抗原抗体结合能力,如天然抗体的组件F(ab)2、F(ab)2'、Fab和Fv片段等(图2)。另外,通过抗体工程改造获得的scFv以及Diabody等则有更好的稳定性和靶向效率。还有一类缩小版抗体则是人源化的非典型抗体片段,如来源于骆驼和羊驼的VHH片段,以及来源于鲨鱼的VNAR片段等,这一类通常被称为纳米抗体。这3种缩小版的抗体都已被用于探索开发缩小版ADC。在天然抗体片段中,利用Fab来设计ADC分子的研究较多。Fab片段包括抗体的单侧抗原结合臂,由抗体的轻链和重链可变区加CH1区段组成,分子质量约为50 ku,可通过酶解完整抗体或者重组表达获得[5。早期的 Fab-ADC采用中等活性的化疗药物紫杉醇和阿霉素作为毒素小分子,其肿瘤抑制活性达不到常规ADC的水平。直到Badescu及其同事利用赫赛汀的Fab片段与MMAE偶联得到的ADC,在体外细胞实验中展示出了亚纳摩尔级别的活性,与常规ADC相当。但是在体内动物实验中,该ADC在剂量高达20 mg/kg时才展现出了中等程度的肿瘤抑制作用,与常规ADC的1~10 mg/kg的有效肿瘤控制剂量相差甚远,分析原因可能是因为缺少Fc-FcRn循环和较高的泌尿系统清除率导致整体肿瘤暴露量低于常规ADC,所以整体抗肿瘤活性达不到常规ADC的水平。其他一些以Fab为基础的ADC分子,有的在体外活性方面就低于常规ADC,有些则是与Badescu研究组的结果一样,在体内活性方面不如常规ADC,因此目前工业界很少有此类ADC进入临床研究。以scFv及其衍生体作为载体则是缩小版ADC的另一个研究方向,scFv由抗体的轻链和重链的可变区通过一段短肽或者二硫键链接而成,其分子质量约为30 ku,且容易在原核细胞中表达,scFv类的ADC已在多个临床前研究中显示出良好的可开发潜力,在采用特殊的重轻链连接方式后,可以通过表面赖氨酸偶联高达10个毒素小分子,在偶联奥瑞他汀(Auristatin)和美登素(Maytansine)后展现出与常规ADC类似的体外肿瘤细胞杀伤活性,同时其肿瘤穿透力也远高于常规ADC。英国的生物技术公司Antikor的研究显示,他们的scFv-ADC比常规ADC有更好的耐受性和安全性,且比Fab等其他抗体片段ADC,其系统清除慢很多,与采用白蛋白辅助延长半衰期手段的抗体片段类似,目前Antikor公司的scFv ADC产品正在临床前开发中。全球ADC研发先驱者Seagen公司曾经采用抗CD30的Diabody制备ADC并与常规ADC进行头对头对比研究,结果显示虽然其药效仅为常规ADC的1/3,但是这是在其血液暴露量仅为常规ADC 1/30的情况下产生的。因此,如果通过诸如聚乙二醇化等方法降低其系统清除率,Diabody ADC有可能会比常规ADC有更好的效果。此外,另一种具备抗原结合能力的抗体Fc片段(Fcab)介导的ADC也展现出了与常规ADC相当的体外肿瘤细胞杀伤活性,同时由于其分子质量仅为抗体的1/3,使得它具备更好的肿瘤穿透力,同时它还具备完整的Fc功能,使得它的半衰期较一般抗体片段长(小鼠血液半衰期为60~85 h),因此具备一定的开发潜力。另外,采用纳米抗体的ADC也展现出了良好的体外和体内肿瘤杀伤作用,如Crescendo Biologics公司的CB108,就是一种采用人的重链可变区的靶向PSMA(prostate-specific membrane antigen)的纳米抗体ADC,展现出了良好的体内疗效和肿瘤穿透性;同时它还包含有抗白蛋白的结合域,通过与血清白蛋白的结合来延长其半衰期,同时还保留其高肿瘤穿透性。另一家公司Elasmogen也正采用类似的纳米抗体在开发ADC,其纳米抗体来源于鲨鱼的抗体可变区。
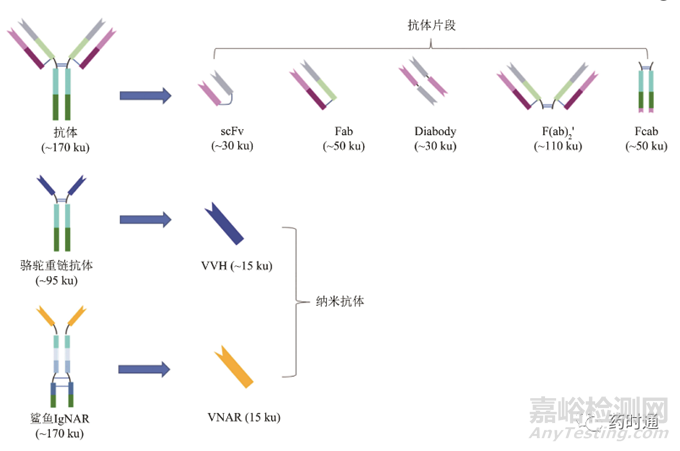
图2 用于ADC药物的抗体片段和纳米抗体结构示意图
Fig. 2 Structure illustration of IgG fragments and nanobodies utilized in ADCs
2、链接体
链接体负责将ADC的抗体和细胞毒小分子链接在一起,对于ADC药物的安全性和稳定性至关重要。理想的链接体需要在血液循环中足够稳定,能有效避免ADC药物在血液循环中和正常组织中释放毒素小分子并使之保持稳定且非活性状态,而同时又需要能在肿瘤组织和细胞内高效释放毒素小分子。另外由于毒素分子通常为疏水性强的化学小分子,链接体需要具有良好的亲水性来连接毒素分子以便于后续在水相中与抗体进行偶联反应并避免大量蛋白聚集体的产生而导致ADC的产率下降。目前常见的ADC链接体有两类:不可剪切链接体和可剪切链接体,它们各有各的特点。近几年,这两类链接体通过不断的改进和优化,开发出了各种能显著提高ADC稳定性和亲水性的链接体。
链接体一般包括4个分子片段:抗体连接片段、调节片段、酶降解片段和自裂解片段(表2)。开发者通常依据毒素小分子的特性以及与抗体连接的方式,并结合对ADC分子的影响,开展综合分析评估来筛选上述4个片段,最终确定链接体的结构。
链接体的稳定性决定了ADC药物中的毒素小分子是传递到肿瘤细胞内部还是有可能在运输过程中过早释放并跟随血浆传递到所有组织,显著影响ADC药物的安全性和有效性。ADC药物的稳定性一般受抗体与链接体的偶联方式、调节片段和氨基酸片段3个方面的影响,其中,偶联方式由抗体连接片段决定,常见连接片段的特点及结构举例见表3。
除连接片段外,调节片段和氨基酸片段的结构也对链接体稳定性有影响,特别是构建氨基酸片段的氨基酸种类和序列,对链接体的稳定性甚至有决定性的作用。2021年,李卓荣课题组报道了不同链接体与MMAE构建的链接体-药物,发现在氨基酸片段为缬氨酸-瓜氨酸(VC)时,调节片段为环己基组成的链接体比起正己基或苯基组成的链接体更稳定,而在调节片段为环己基时,氨基酸片段为甘氨酸-苯丙氨酸-亮氨酸-甘氨酸(GFLG)组成的链接体稳定性要远低于其他氨基酸片段组成的链接体。
在调节片段中引入磺酸或聚乙二醇等极性基团,既能够增强链接体-药物的水溶性,降低制备ADC分子时产生聚集体的几率,又能够增强毒素小分子克服P糖蛋白(P-glycoprotein,Pgp)外排效应的能力。Kovtun等研究了分别包括环己基和四聚乙二醇两个调节片段的链接体在ADC中的应用,发现使用四聚乙二醇片段的ADC在具有多药耐药性(multi-drug resistance)细胞系COLO 205MDR CDX模型中具有更好的细胞杀伤效应,即使给药剂量降低1倍,也能够有效地抑制肿瘤细胞生长。
总体而言,链接体的设计对于ADC来说至关重要。近些年链接体技术方面也有一些新的扩展,例如Bolt公司的BDC-1001和Silverback公司的SBT6050及SBT6290等多款免疫激动剂抗体偶联物(ISAC)所采用的在肿瘤微环境释放的一类链接体,该类ADC分子通过抗体与肿瘤细胞表面非内吞靶标或肿瘤微环境中的特异性靶点结合,然后在肿瘤细胞外释放毒素小分子,利用毒素小分子的旁杀效应或激活肿瘤组织浸润的免疫细胞,达到抑制肿瘤细胞增殖的作用。
与常规ADC相比,非内吞ADC对链接体的要求有着显著的区别(表4),需要能够在肿瘤微环境中通过不同的方式有效释放载荷分子,而不是在细胞内的溶酶体中。
3、毒素小分子
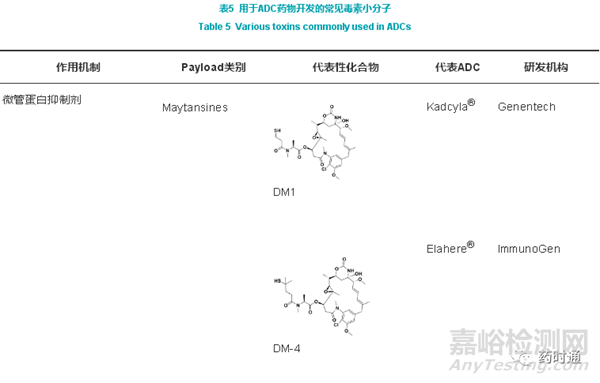
毒素小分子是ADC药物中主要负责执行细胞杀伤功能的组件,它需要具备较高的活性使之能在较低的剂量下有效杀伤肿瘤细胞。目前主流的ADC药物其毒素小分子的细胞抑制的IC50基本在10-9~10-12 mol/L范围之间。同时,它还需要具备很好的稳定性,使之在循环系统和溶酶体中保持结构完整和活性。另外,它还需要分子质量足够小,免疫原性低,也需具备方便与链接体进行化学反应的位点。目前常见的毒素小分子主要包括微管蛋白抑制剂和DNA复制的抑制剂,随着日本第一三共公司的重磅ADC药物Enhertu®应用拓扑异构酶抑制剂的成功,越来越多的细胞毒性相对较低的小分子进入到ADC药物开发者的工具箱,如针对拓扑异构酶的喜树碱类、RNA聚合酶抑制剂、细胞凋亡调控药物等,常见毒素小分子汇总于表5。
3.1 微管蛋白抑制剂
微管蛋白作为细胞骨架的主要构成物质,除能够支撑细胞结构完整性外,同时还在细胞增殖的有丝分裂阶段具有至关重要的作用。通过抑制微管蛋白的生成和聚集(polymerization),既能够杀伤肿瘤细胞,也可以抑制肿瘤细胞的快速增殖。常用于ADC构建的微管蛋白抑制剂包括美登素(Maytansines)、海兔毒素(Auristatins)和Tubulysins等。
现阶段,已进入商业化的14款ADC药物中,有8款所使用的毒素小分子是该类化合物,其中5款使用MMAE(海兔毒素衍生物)、1款使用MMAF(海兔毒素衍生物)、1款使用DM1(美登素衍生物)和1款使用DM4(美登素衍生物)。微管蛋白抑制剂类化合物的特点是:杀伤细胞活性高,IC50一般在10-9~10-10 mol/L之间;具有一定的亲水性,不易引起大分子聚集;起效速度快,短时间内即可杀伤肿瘤细胞。虽然MMAE应用广泛,但因其是Pgp底物而存在多药耐药性的问题。
而Tubulysins类化合物作为微管蛋白抑制剂则不是Pgp底物。该类化合物的结构均具有官能团——羧基,这一亲水性基团增加了毒素小分子的极性进而规避了Pgp的外排效应,毒素在随着ADC分子进入肿瘤细胞后起到杀伤效力,但同时,由于细胞膜穿透能力的降低,无法扩散至周围肿瘤组织,因此也就不具备旁杀效应。
3.2 DNA损伤剂
DNA损伤剂因作用机制不同而分为3大类:DNA双链破坏剂、DNA插入剂和DNA烷基化剂。DNA在细胞的生长和增殖过程中至为关键,对其进行破坏,能够高效杀伤肿瘤细胞并抑制其快速增殖。值得注意的是,部分DNA损伤剂,如PBD dimer和Duocarmycins,在动物中高剂量给药时,可能产生延迟毒性,因此在给药周期设计时,应谨慎考虑。
3.2.1 DNA双链破坏剂
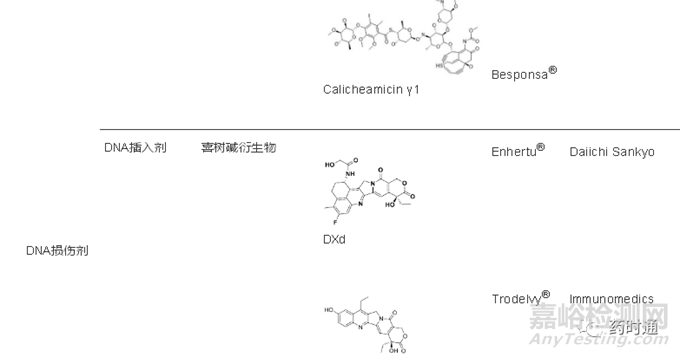
DNA双链破坏剂的化学结构中包含一段烯二炔片段,进入细胞核后,该片段发生Bergman环化反应,形成苯环双自由基过渡态,诱导DNA双键断裂。此类毒素小分子的代表是卡利霉素衍生物γ1(calicheamicin gamma 1),辉瑞公司的2款商业化ADC药物Mylotarg®和Besponsa®均使用该类化合物。
3.2.2 DNA插入剂
在DNA复制和转录阶段,拓扑异构酶I发挥着重要的作用,它以共价键的形式与DNA形成一个可裂解的复合物,从而产生一个单链缺口,另一条未受损的单链从缺口中回转,使超螺旋的DNA松弛,以利于复制和转录,当解旋完成后拓扑异构 酶I脱离并促使DNA链复原。因此,如果能在拓扑异构酶I与DNA形成可裂解复合物的阶段引入一个分子,能够阻挡未受损单链的回转,就可以阻止DNA的复制和转录,诱导肿瘤细胞死亡。喜树碱就是这样一类拓扑异构酶I抑制剂类化合物,目前已有2款商业化ADC药物Enhertu®和Trodelvy®使用该类化合物作为毒素小分子。喜树碱类化合物的细胞杀伤活性相较于微管蛋白抑制剂弱1~2个数量级,能够扩展ADC药物的治疗窗,提高给药剂量。
3.2.3 DNA烷基化剂
DNA烷基化剂一般能够与DNA小沟结合,之后,利用分子内易与氨基反应的官能团,如碳氮双键或环丙烷结构,与鸟嘌呤或腺嘌呤发生亲核反应,在DNA中形成链间或链内交联,阻止其复制和转录,导致肿瘤细胞死亡。
该类化合物细胞杀伤活性在皮摩尔级别,比微管蛋白抑制剂活性更强,因此,能够大幅降低ADC的给药剂量,但治疗窗也会相应缩小。常用的DNA烷基化试剂为PBD(pyrrolobenzodiazepine)二聚体和Duocarmycin两类化合物,目前已进入商业化的ADC中仅有一款使用PBD二聚体作为毒素小分子——CD19靶向的Zynlonta®。
3.3 其他毒素小分子
除上述主要已应用于商业化ADC药物的毒素小分子外,其他各类作用机制的毒素也被逐步应用于ADC药物开发中,如Bcl-xL抑制剂、RNA剪切酶抑制剂、RNA聚合酶抑制剂等。另一类新颖毒素小分子则作用于细胞膜表面靶标或者肿瘤组织基质靶标,用于非内吞型ADC药物的开发,如NKA(Na+/K+-ATPase,钠钾离子泵ATP酶)抑制剂、基质金属依赖性蛋白酶(matrix metalloproteinases,MMP)抑制剂和免疫激动剂等,其各自的作用机制如表6所示。
4、偶联技术
ADC药物开发成功的另一个重要元素是将小分子组分(链接体或链接体-毒素小分子)加载在抗体分子上的方式和技术。偶联反应策略和过程,决定了载药量(drug antibody ratio,DAR)和载药分布方式等关键质量属性,与ADC药物的有效性和安全性直接相关。理想的ADC偶联策略或技术需具备如下特点:a. 抗体与小分子结合部分的化学键或基团应足够稳定,确保其在循环系统中的稳定性;b. 偶联位点不会干扰抗体的功能,特别是与靶点抗原结合的特异性和高亲和力;c. 偶联过程涉及的反应必须有足够高的选择性和反应效率,同时应易于控制载药量和载药分布。
目前的ADC偶联技术大致可以分为两种类别,一种是利用抗体序列中天然的具备反应活性的氨基酸残基(如表面赖氨酸的侧链氨基和链间二硫键还原后的巯基)来介导的偶联技术,目前已上市的13款ADC药物采用此类偶联技术;另一类技术,则是通过化学修饰、基因工程技术或者酶修饰等手段在抗体特定位点引入可供反应的基团,再偶联上毒素小分子,实现特定位点偶联。此类技术主要包括工程化半胱氨酸定点插入、非天然氨基酸定点插入、酶介导以及N-糖链介导的偶联技术等。目前采用定点偶联的ADC药物均处于临床前或临床研究阶段。
4.1 表面赖氨酸侧链氨基介导的偶联技术
赖氨酸残基的一些特性使其成为蛋白质生物偶联的首选:a. 在蛋白质包括抗体中广泛存在;b. 在蛋白质表面分布较多且易于接触;c. 其侧链ε位氨基具备良好的化学反应活性。利用具备反应活性的羧酸酯基团,如氮-羟基琥珀酰亚胺(NHS)的链接体,即可与赖氨酸的ε位氨基反应形成共价键实现小分子偶联,操作过程简单易行。这些优点使得它成为蛋白质标记的常用手段,早期的ADC药物也采用此类偶联技术,如Mylotarg®和Besponsa®,而且后来的Kadcyla®和2022年获批的Elahere®也是采用此技术。
由于IgG分子上大约有80~90个赖氨酸残基,其中有约一半是可以被修饰的,此种偶联技术虽然可以通过控制反应条件来控制平均DAR值,但是控制偶联位点就很困难,而导致其单个抗体上可能出现的偶联位点组合如果按照完全随机的理论计算可高达数百万种,此类ADC药物的载药量分布不均一,成为由不同载药量的偶联物亚群组成的混合物,而不同的载药量,会对ADC的药效和药代动力学特性产生影响。另外,由于赖氨酸分布在整个抗体表面,偶联如果发生在抗原抗体结合域,就有可能干扰ADC与靶点抗原的结合能力。尽管此技术存在天生的产生不均一性偶联的特点,近些年对Kadcyla®(T-DM1)结构的深入研究显示,通过合理的工艺设计和深度工艺开发,在合理的工艺控制条件下,仍能将批次间以及不同生产规模的ADC产物中偶联位点分布和载药量保持高度一致,这也就是应用该技术的Mylotarg®、Besponsa®、Kadcyla®和Elahere®能开发成功的关键之一。采用此技术的ADC会根据链接体和毒素小分子的特性以及工艺的可控性和收率等,去选择使用一步法或者两步法来合成ADC分子(图3a)。一步法是先将链接体和毒素小分子连接,再通过链接体上的反应基团与抗体表面的赖氨酸侧链氨基偶联;而两步法则使用具备双功能基团的链接体上的一个反应基团与抗体上赖氨酸侧链氨基偶联完成抗体修饰,之后链接体上的另一个功能基团再与毒素小分子偶联制成ADC分子。
4.2 抗体链间二硫键介导的偶联技术
半胱氨酸残基由于其还原状态的巯基具备极强的亲核性,使它成为另一个特殊属性的生物偶联媒介。以IgG1为例,它总共含有16对二硫键,其中12对为链内二硫键包裹在其结构内部而不易修饰,而4对链间二硫键则暴露于溶液中可以被还原剂打开(图3b),从而产生最多8个可供反应的自由巯基。虽然通过控制还原剂的比例可以很好地控制产物的平均DAR值,但是除了完全打开4对二硫键并全部偶联上毒素小分子的ADC药物(如Enhertu®和二硫键桥接技术介导的ADC),其他通过部分还原和偶联的ADC其载药量分布依然是非均一的。不过得益于有限数量的巯基,其异质性要远低于赖氨酸介导的偶联,主要的载药量组分(isomer)是载有0、2、4、6、8个小分子的组分。利用巯基的偶联反应,通常通过与氮取代的马来酰胺基团产生1,4位的Michael加成反应来实现,这个反应速度快且反应条件温和,很适用于偶联反应。
有研究表明采用此类偶联技术的ADC可能会由于打开抗体的链间二硫键而导致抗体的完整性和稳定性受到影响,但是Seagen公司的科学家们通过对CD30靶向的Adcetris®药物分子深入的结构研究发现,尽管将IgG1抗体的四对链间二硫键都打开,偶联上8个MMAE毒素小分子对抗体的高级结构仅引起微小的变化,而且这些变化对ADC的整体稳定性并无实际意义的影响,当然这种偶联方式是否会对抗体的完整性和稳定性造成影响可能不能一概而论,而是需要对各自的ADC药物分子及其抗体在偶联前后进行仔细的结构分析对比。此类偶联技术的另一个需要关注的点是,其偶联化学基团巯基琥珀酰亚胺有发生反Michael加成反应的倾向,从而使所偶联的小分子转移至血清中带巯基的蛋白质上,影响其血浆中稳定性。但是,抗体链间二硫键介导的偶联技术对比其他技术,有着许多明显的优点,比如其生产工艺相对简单,反应条件易于控制以及得率高等,使之成为目前ADC药物开发中应用最为广泛的技术。目前已上市的14款ADC药物中有9款采用此技术。
另外,利用抗体链间二硫键介导的偶联也能实现定点偶联,如将IgG1的全部8个经过链间二硫键还原后的巯基都偶联上毒素小分子的Enhertu®,还有就是链间二硫键桥接技术。
二硫键桥接技术通过还原剂将IgG1的4对链间二硫键完全打开,再采用带有两个可与巯基反应基团的叉状双接头,同时与打开的一对链间二硫键反应,将打开的二硫键通过链接体上的双接头重新链接起来,实现一对链间二硫键一个链接体分子,同时恢复了抗体的链间连接和结构稳定性(图3c)。而载药量则由每个链接体上携带的细胞毒分子数量所决定,可以为4、8或16。此类技术无需基因工程改造,使用天然的链间二硫键就可以实现,但是需要设计合适的携带双巯基反应基团的链接体。目前有多种二硫键桥接定点偶联技术,如双砜(bis-sulfones)技术、下一代马来酰胺(next-generation maleimides,NGMs)技术、马来酰肼(pyridazinediones,PDs)技术以及 C-LockTM技术,都已被用于ADC药物开发和探索中。采用双砜技术偶联产生的符合预期的一对链间二硫键一个链接体分子的组分大约占比75%~85%,需要进一步使用疏水层析纯化,才可使目标组分达到95%以上。OBI Pharma公司运用双砜技术将MMAE偶联至抗-GloboH的抗体上开发出ADC药物候选分子OBI-999,用于治疗胰腺癌和胃癌,目前正在临床研究中。也正是在对此分子的结构研究中发现这一ADC中存在两种异构体,一种是完全按照天然抗体二硫键重新桥接的方式,另一种则是一种“半抗体形式”,即重链铰链区的二硫键经桥接后形成了链内桥接形式,而两个重链间则是分离状态,无共价键结合,目前尚不知此异构体是否会影响药效。而NGMs技术偶联产物,也与双砜技术一样,存在两种偶联异构体,一种是天然抗体形式,另一种则是“半抗体形式”,同样也会跟常规的马来酰胺一样在血液中发生巯基交换反应而导致小分子提前释放,目前此类偶联技术的ADC尚未进入临床研究中。马来酰肼技术比起前两种技术的优势在于,其制成的偶联产物血清稳定性较好,而且此类偶联物目标产物——天然二硫键桥接形式的ADC占比可达90%以上,因此被认为是目前最佳的二硫键桥接方式之一。而 C-LockTM技术则是由Concortis Biotherapeutics所开发,其代表作为Sorrento Therapeutics 所开发的STI-6129和Zova Biotherapetics所开发的ZV0508,分别靶向CD38和5T4,毒素小分子均为多司他汀(Duostatin)。二硫键桥接技术目前仍在不断推陈出新,期望能解决偶联桥接导致的“半抗体”异构体问题,改善血清稳定性,提高此类ADC的安全性和有效性。
4.3 工程化半胱氨酸定点偶联技术
工程化半胱氨酸偶联技术是在抗体表面指定位点引入半胱氨酸而实现定点和均一性偶联的一类技术(图3d),如Genentech公司的THIOMABTM技术。在一份发表的研究中,通过基因工程技术将半胱氨酸通过点突变的形式引入到一个抗MUC16靶点的单抗重链114位(A144C),通过CHO细胞表达纯化后,经过TCEP部分还原将114位的半胱氨酸的巯基暴露出来,再以硫酸铜氧化被打开的链间二硫键,最后再与携带有马来酰胺基团的小分子vcMMAE反应,所产生的ADC药物平均DAR值为1.6。THIOMABTM ADC与通过链间二硫键介导的DAR值为3.1的ADC相比,尽管载药偏低但是其体内动物模型药效并无区别,且展现出了更好的安全性,同时血清清除率要远低于非均一ADC,而且THIOMABTM ADC经过工艺优化后可以达到平均DAR为2,产物中DAR2的组分占比超过90%。THIOMABTM和其他一些类似技术在进行偶联前,均需要通过还原去除抗体在细胞培养过程中所引入的半胱氨酸上的配基。有意思的是,一篇文献报道,在赫赛汀的轻链124位(Q124C)突变引入半胱氨酸,这种抗体表达后纯化时其124位半胱氨酸由于空间位阻的原因而没有和配基结合,因此可以直接用于偶联反应,而且其偶联产物均一,DAR值为2,这一发现对于进一步优化THIOMABTM技术,简化生产工艺有所启迪。另外还有一些与THIOMABTM类似的技术,如通过基因工程在特定位点插入半胱氨酸而不是由点突变获得,还有在多个位点同时引入半胱氨酸以增加载药量的尝试。该技术已在体内外临床前研究中展示了其疗效和安全性,目前已有使用此类技术的ADC药物进入临床研究中。近期,辉瑞公司研究发现了一种新型的制备此类ADC的生产技术,避免了先完全还原后氧化工艺,采用一步法实现定点偶联,简化工艺的同时降低了还原氧化过程产生的抗体二硫键错配等风险。他们运用一种含有Ellman试剂DTNB(5,5'-二硫双(2-硝基苯甲酸))的培养基来表达生产含有工程化半胱氨酸突变的抗体,使得生产出来的抗体其半胱氨酸上的巯基保护基团为TNB而不是半胱氨酸或谷胱甘肽。此TNB与半胱氨酸形成的二硫键,能被具备温和还原能力的还原剂TSPP(三苯基膦三间磺酸盐)还原,而同时抗体的链间二硫键依然保持稳定,还原产生的巯基与链接体-毒素小分子偶联即可形成定点偶联ADC。此技术产出的ADC药效与采用常规THIOMABTM技术的ADC等同,却不含有THIOMABTM技术制成的ADC中常见的二硫键错配异构体以及片段。
在开发工程化半胱氨酸偶联技术时,半胱氨酸引入的位点选择对于ADC的药代动力学特性和毒副作用都有影响,因此需要谨慎筛选合适的位点来进行基因工程改造和偶联。如Ma等在一个人源化抗IL13Rα2(interleukin 13 receptor subunit alpha 2)抗体的恒定区引入了一系列的半胱氨酸突变,然后利用这些工程化的抗体制备ADC,之后在大鼠和小鼠中对这些ADC进行了药效、药代动力学和毒理研究。结果显示,所有的定点偶联ADC都比常规ADC有更好的体内药效和耐受性,而不同位点偶联的ADC之间则展现了出了不同的血清清除率和脱靶毒性,某些位点偶联的ADC(L443C)比起其他的具备更低毒副作用和更长的半衰期。
4.4 非天然氨基酸定点偶联技术
非天然氨基酸定点偶联技术的核心是通过基因工程技术以及对表达体系的tRNA编码改造,将带有反应活性基团的非天然氨基酸引入到抗体的预定位置,从而介导定点偶联(图3e)。此类技术比较常用的非天然氨基酸有对乙酰苯丙氨酸、叠氮取代甲基苯丙氨酸和叠氮取代型赖氨酸。运用此类技术开发的ADC分子,已被证明具备高均一性的载药量和载药分布,且具备优秀的活性和稳定性,其安全性也很好,因此吸引了很多学术界和医药生物技术公司对此类技术进行探索和开发。目前采用此类技术的代表性ADC药物是Ambrx和浙江医药联合开发的ARX788,ARX788的靶点是HER2,运用的非天然氨基酸是对乙酰苯丙氨酸,通过肟键与链接体药物结合,采用的链接体为不可剪切的聚乙二醇,毒素小分子为Amberstatin,平均载药量为1.9。此外,生物技术公司Sutro也有两个采用此类技术的ADC药物分子处于临床研究中,分别是STRO-001和STRO-002,它们运用的非天然氨基酸为叠氮取代甲基苯丙氨酸,通过点击化学(click-chemistry)反应完成偶联。STRO-001是以抗CD74抗体为载体通过一种不可剪切的链接体与美登素偶联而成,平均载药量为2,临床研究的适应症为DLBCL。STRO-2则是靶向叶酸受体的ADC,运用可剪切VC(valine-citrulline)链接体及微管抑制剂SC209为毒素小分子,平均载药量为4,临床I期研究的结果显示此ADC具有良好的耐受性、可控的副反应,以及治疗卵巢癌的潜力。除了这些ADC分子外,还有其他一些采用非天然氨基酸定点偶联的ADC药物处于临床前研究中,而且采用此类技术开发ADC的项目仍在快速增加中。但是此类ADC技术的缺点也很明显,携带非天然氨基酸的抗体表达量通常偏低,成本较高,且引入非天然氨基酸可能会产生免疫原性增高的风险。
4.5 酶介导的定点偶联技术
酶类由于具备很好的位点或氨基酸序列选择性,且其最佳活性条件与抗体的舒适条件一致,因此也被广泛用于介导定点偶联。酶类或者通过直接将小分子药物转移至特定氨基酸序列上,或者通过将含反应基团的链接体引入到抗体特定位点从而达成定点偶联(图3f)。目前比较常见的用于定点偶联的酶有谷氨酰胺转移酶(transglutaminase,TG)和甲酰甘氨酸生成酶(formylglycine-generating enzyme,FGE)。谷氨酰胺转移酶能特异性介导蛋白质中谷氨酰胺末端的甲酰胺与赖氨酸上的侧链氨基反应形成共价键,其中微生物来源的谷氨酰胺转移酶(mTG)在介导偶联方面最为常用。mTG介导的定点偶联策略有4种:第一种采用基因工程技术或糖苷酶解将抗体上糖苷切除,使295位上的谷氨酰胺暴露以供mTG修饰;第二种是运用基因工程技术将mTG特异识别序列(如LLQG等)添加到重链或轻链的C端以供mTG定点偶联;第三种则可直接利用抗体末端赖氨酸残基或者利用基因工程在末端引入赖氨酸残基再利用mTG来修饰;最后一种是将这几种方法组合,以达到增加偶联位点和载药量的目的。在将修饰基团引入特定位点后,可以采用前述的各种偶联化学反应,如click反应和Michael加成反应等,实现毒素小分子的定点偶联。采用mTG修饰的ADC目前均在研究探索阶段,一些研究显示对比常规ADC,其在动物模型中的肿瘤抑制活性更好且持续时间更长。
甲酰甘氨酸生成酶(FGE)能将特定序列CXPXR(X可为任意氨基酸除了脯氨酸)中的半胱氨酸残基进行氧化生产甲酰甘氨酸,甲酰甘氨酸上的醛基可与带有肼或者羟胺基团的小分子药物偶联。这一技术被称为SMARTag®技术,它通过将SMARTag®(CXPXR)整合至抗体的特定位置,如轻链或重链的C端,接着以FGE处理产生反应基团,介导定点偶联。利用这一技术的ADC目前有一款进入了临床研究阶段,其靶标为CD22,在抗体两个重链C端连接有SMARTag®,通过不可剪切链接体与美登素偶联,在临床前和I期临床试验中均展现出良好的疗效和安全性。
4.6 糖链介导的定点偶联技术
在所有抗体的重链CH2区域都有一个保守的N-糖链位点,N-297。利用N-糖链来介导偶联具有先天优势。首先N-糖链的位点远离抗体的抗原抗体结合区,此处偶联一般不会干扰其抗原结合能力;其次,N-糖基化的方式在各种抗体都十分保守,利于糖链修饰技术在不同种抗体中的应用,且无需基因工程改造,使用天然抗体即可;再者就是糖链是由碳水化合物组成,理化性质与蛋白质和氨基酸截然不同,因此从化学反应的角度利于偶联位点的特异性。糖链定点偶联技术主要有通过氧化修饰和糖苷酶修饰两种途径。糖苷氧化修饰技术主要用于抗体标记,通过高浓度氧化剂作用于糖链末端的糖苷的二醇基团使之产生醛基,再用含肟或者羟胺基团的链接体小分子与之反应,实现定点偶联。由于期间需要使用高浓度的氧化剂,如NaIO4,对抗体的氨基酸残基的破坏会导致理化性质甚至药代动力学特性发生改变,因此很少用于ADC药物的开发。目前用于ADC药物开发的主流的糖链偶联技术大都采用糖苷酶技术,其中最为成熟的当属荷兰Synaffix公司开发的GlycoConnectTM技术,目前已授权全球多家ADC药物开发企业,多个产品也正在开展临床研究。该技术首先使用糖苷内切酶将糖链切除只剩余最核心的氮乙酰葡萄糖胺,再使用糖苷转移酶将携带有反应基团的糖苷衍生物(如叠氮取代的乙酰半乳糖胺)结合至核心糖苷上完成定点修饰,最后以click化学反应与链接体药物反应,达成糖链介导的定点偶联(图3g)。采用GlycoConnectTM技术的ADC有两个偶联位点,其载药量取决于每个链接体携带的小分子药物的数量,其修饰和偶联效率可以达到90%以上,在体内和体外实验中展现出优于常规ADC的药效和循环系统稳定性。另外还有一些糖苷酶介导的偶联技术,其技术路线与GlycoConnectTM相似,只是将携带反应基团的糖苷更换为糖链,又或者采用半乳糖苷酶将糖链末端的半乳糖切除,再以转移酶将糖苷或糖链衍生物连接到抗体糖链末端;还有一种就是在抗体表达过程中用带反应基团的岩藻糖衍生物替换天然的岩藻糖,将其引入到糖链岩藻糖的位点以实现定点修饰和偶联。糖链定点偶联技术由于上述的天然优势,使之成为定点偶联技术的重要发展方向。然而,这类技术也有其相应的限制,如需要特殊的试剂和糖苷酶,而且由于偶联位点只有两个,因此其载药量选择较少,而且糖链被修饰后对ADC整体药效和药代动力学的影响还需要进一步研究和评估。
5、ADC药物未来发展方向
从第一个ADC药物在2000年获批上市到现在的20多年间,共计14款ADC药物获批上市,这期间ADC药物已经在偶联技术、链接体和毒素小分子方面取得显著发展,使得其疗效和安全性得到了很大的提高,也吸引了学术界和工业界对该领域的持续关注和研究投入,同时使得ADC技术得到了更进一步的快速发展,为未来ADC药物的设计和开发提供了广阔的前景。
在ADC药物的靶点选择方面,除了肿瘤细胞表面靶点外,肿瘤微环境的靶点是该领域的新热点之一。此类靶点通常包括肿瘤微环境(tumor microenvironment,TME)中的表皮细胞和成纤维细胞抗原。此类靶点,因其分布在肿瘤组织周围,比肿瘤组织更容易从血液循环接触到ADC分子;再者,此类组织细胞比起肿瘤细胞受到基因突变影响小的多,因此产生耐药性的可能性也降低;作用于此类靶点除了能运用毒素小分子杀伤肿瘤细胞外,还可能产生抗血管生成等协同抑制肿瘤增殖的作用;最重要一点,TME抗原很少或者几乎不在正常组织表达。LRRC15(leucine-rich repeat containing 15)就是此类靶点的代表,它在多种肿瘤的微环境里的成纤维细胞膜表面有高表达,而在正常组织不表达。艾伯维公司的ABBV-085则是针对LRRC15开发的新型ADC,由抗LRRC15抗体和MMAE偶联而成,在含有LRRC15阳性基质的肿瘤中展现了良好的活性和安全性。另一种肿瘤基质靶点的ADC,则同时运用了另一种新型毒素小分子释放机制——非内吞依赖性的释放,如靶向肿瘤组织中的不溶性纤维蛋白(insoluble fibrin,IF)的一种ADC,运用抗IF抗体和MMAE经血纤维蛋白溶酶特异性剪切的肽段偶联而成,动物实验研究结果显示它仅在与IF结合后释放MMAE,而释放的MMAE除了杀伤肿瘤细胞外还能破坏肿瘤中的血管,起到协同杀伤作用,展示了其开发潜力。
在ADC药物的靶点选择方面,除了肿瘤细胞表面靶点外,肿瘤微环境的靶点是该领域的新热点之一。此类靶点通常包括肿瘤微环境(tumor microenvironment,TME)中的表皮细胞和成纤维细胞抗原。此类靶点,因其分布在肿瘤组织周围,比肿瘤组织更容易从血液循环接触到ADC分子;再者,此类组织细胞比起肿瘤细胞受到基因突变影响小的多,因此产生耐药性的可能性也降低;作用于此类靶点除了能运用毒素小分子杀伤肿瘤细胞外,还可能产生抗血管生成等协同抑制肿瘤增殖的作用;最重要一点,TME抗原很少或者几乎不在正常组织表达。LRRC15(leucine-rich repeat containing 15)就是此类靶点的代表,它在多种肿瘤的微环境里的成纤维细胞膜表面有高表达,而在正常组织不表达。艾伯维公司的ABBV-085则是针对LRRC15开发的新型ADC,由抗LRRC15抗体和MMAE偶联而成,在含有LRRC15阳性基质的肿瘤中展现了良好的活性和安全性。另一种肿瘤基质靶点的ADC,则同时运用了另一种新型毒素小分子释放机制——非内吞依赖性的释放,如靶向肿瘤组织中的不溶性纤维蛋白(insoluble fibrin,IF)的一种ADC,运用抗IF抗体和MMAE经血纤维蛋白溶酶特异性剪切的肽段偶联而成,动物实验研究结果显示它仅在与IF结合后释放MMAE,而释放的MMAE除了杀伤肿瘤细胞外还能破坏肿瘤中的血管,起到协同杀伤作用,展示了其开发潜力。
由于目前限制ADC效力的一个重要因素是其分子尺寸太大,使之不容易穿透毛细血管以及肿瘤基质等屏障到达肿瘤细胞,从而阻碍了其在实体瘤治疗方面的效果。因此,针对这一局限性,研究者们尝试了各种手段缩小ADC尺寸,包括前述的抗体片段或纳米抗体等形式的ADC。此外,还有一类利用更小尺寸非抗体框架的ADC,其抗体部分由一段有靶向结合能力的多肽替代以进一步增加其穿透力,这一多肽由化学合成得到。目前已报道的最小分子质量的一类多肽框架ADC,是一类被称为双环多肽(bicyclic peptide)的肽段,分子质量仅1.5~2 ku,其次是一类被称为“Pentarin”的分子,分子质量为2~5 ku,二者分别由生物技术公司Bicycle Therapeutics和Tarveda公司所开发的技术,其代表性ADC产品分别是BT5528和PEN-221。BT5528的靶点为EphA2(Ephrin A2)受体,由靶向EphA2的双环多肽与MMAE偶联而成,其与靶点的亲和力 1.9 nmol/L。小鼠荷瘤实验研究结果显示,它能快速被肿瘤吸收且能持续地聚集在肿瘤组织中。而且临床前毒理实验显示,它没有观察到同靶点常规ADC MEDI-547的相关毒性,该分子目前在临床研究中。而PEN-221目前也在临床研究中,它是由靶向生长激素抑制受体2(SSTR-2)的奥曲肽衍生物与DM1偶联形成,它与靶点的亲和力很高在 51 pmol/L左右。动物实验结果显示1~2 mg/kg的剂量下就能很好地抑制肝肿瘤和肺肿瘤,且在I期临床试验中已展现出其疗效和安全性。此类ADC开发的最大挑战就是其血液清除率太快,将来如能克服此缺点将会进一步拓展其治疗适应症,如脑肿瘤和一些血管分布很少的实体瘤。
另一些尝试主要是在载荷方面,如通过在抗体上偶联两种不同的细胞毒小分子,以应对肿瘤组织中癌细胞的高度异质性和耐药性。Seagen公司的研究者们通过在抗CD30抗体cAC10上同时偶联上MMAE和MMAF,首次在动物实验中展现出了双载荷ADC能有效地杀伤对MMAE-ADC有耐药性的肿瘤细胞。Yamazaki和同事则将此研究更进一步,利用定点偶联技术,以及含多载荷连接位点的链接体,可以调整两种不同细胞毒小分子的比例,构建出MMAE+MMAF分别为2+2、4+2和2+4等组合的HER2靶向ADC分子。动物实验显示,双载荷ADC比起单载荷ADC或者联合使用两种单载荷ADC,针对HER2阳性的杂合型加复发性乳腺癌肿瘤组织有更好的疗效,且不同的载荷比例显示不同的抗肿瘤活性,为将来克服肿瘤的异质性和耐药性提供了一种新的思路。
此外,还有将ADC中传统的细胞毒小分子载荷替换为蛋白毒素、细胞因子、蛋白降解靶向嵌合体(proteolysis-targeting chimeras,PROTAC)和小核酸作为载荷,用于治疗癌症和其他疾病。如Avidity Biosciences的AOC 1001靶向肌肉组织上靶点TfR1(transferrin receptor 1),其siRNA作用于DMPK mRNA,用于治疗一种罕见的肌肉病变。目前这些种类的偶联药物还处于早期研发阶段,有的已经开始进入临床研究阶段,有的才刚刚完成概念验证,都还面临各种各样的挑战,相信将来随着各种技术的进步和成熟,特别是对疾病机理的更深入理解,加上人工智能辅助药物设计技术等,这些新形式的ADC药物会得到进一步的发展,造福更多患者。
6、结论
ADC药物设计的本质是利用抗体的高选择性靶向运输能力将具有生物学效应的小分子药物,如细胞毒小分子药物高效地递送至病灶发挥治疗作用,既是一种新型药物分子,又是一种靶向递送系统。在这一体系内,抗体、链接体、小分子以及偶联技术等关键要素都需要作为一个整体来综合考虑,针对靶点的生物学特征以及疾病的特性和机理,选择合适的组件和技术对于开发成功的ADC药物至关重要。为了进一步发掘ADC药物的潜力,国内外在各个关键要素上都正在进行大量的研究,为开发新一代的ADC药物持续充实工具库,并提供源源不断的新思路。这些新思路包括:选择靶向肿瘤微环境的抗体并结合使用细胞外释放机制的链接体药物,解决靶点相关的耐药性;使用分子质量较小的抗体片段或者纳米抗体开发ADC,提高肿瘤组织穿透力;选择亲水性更好的链接体改善ADC的载药量和理化性质;使用不同作用机制的小分子药物,降低毒副作用并拓展ADC药物的应用领域;选择定点偶联技术改善载药均一性和药代动力学特性等。相信在ADC领域研究者的共同努力下,克服现有ADC的缺点和挑战,下一代的ADC药物将会给肿瘤的靶向治疗带来新惊喜,使更多的肿瘤患者获益。另外,作为一种递送系统,相信将来ADC药物必将用于更加广泛的领域,如中枢神经疾病、遗传疾病和感染性疾病等领域,作为真正高效的“魔法子弹”去消灭疾病,拯救生命。
参考来源:生物化学与生物物理学进展. 刘文超, 李鸿峰 ,胡朝红.