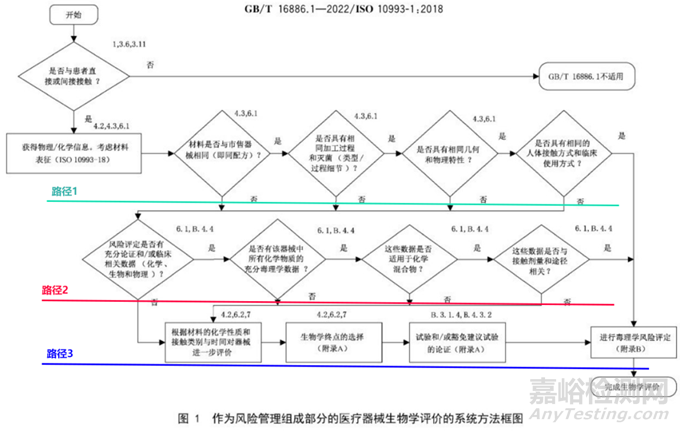
总体而言,根据GB/T 16886.1-2022图1 可以得知,医疗器械的生物学评价可通过以下三种路径进行评价:
路径1为通过与国内市售器械毒理学等同性的方式进行评价
路径2为通过可沥滤物化学表征及毒理学分析的方式进行评价
路径3为通过生物学试验的方式进行评价
后续将分别根据刘老师介绍的内容进行归纳总结提炼。
第一部分:路径1-通过与国内市售器械毒理学等同性的方式进行评价
在开始进行生物学评价之前需要确认否接触,如果不接触,则不适用,无需进行生物学评价。如果接触,则继续往下一个逻辑框进行。首先,获取相应的物理化学信息,然后对材料进行表征。接下来,在获取这些信息后,需要与市售的器械进行等同性比较。
需要说明的是:“市售器械”指的是中国已上市的器械。国外的已上市器械不符合要求,如果说选择一个国外的国内都没有上市的产品,其实它到底是不是符合要求你不知道,所以你选择跟他建立等同性也是没有必要的。因此要证明等同性,需要与在国内上市的器械建立等同性。这个与临床器械选择对照品的原则是一样的。同时在对比中,需要按照逻辑框分别进行。
1.材料(配方)包括组成成分及可沥滤物:
这里刘老师采用茶包泡茶举了一个特别好理解的例子,关于材料的配方组成,我们评价的不单单是茶包,还要更要关注从茶包里边萃取出来的这些物质,就要对这些可萃取的物质来进行系统分析。
2.加工过程和灭菌(考虑加工工艺引入物质及对终产品的影响)
对于加工过程和灭菌方式,因为这些往往会引入相关物质,并且对终产品的生物学风险造成一些影响,所以这也是需要重点考虑的。综合上面几点,其实我们可以看出来,如果一个企业和自家生产的已上市的产品来进行比较,还是具更现实,也具有可操作性。你往往不知道其他家的它生产加工过程,就更别说里边过程细节了。
3.几何和物理特性(表面形态、多孔结构、纵横比、厚度、纳米颗粒、亲疏水、溶胀、耐磨性等)
那么路径一后面的这两个框,一个是是否具有相同的几何和物理特性?这几何物理特性就是前面咱们也提到了,它对于某些生物学终点还是有一个比较大的影响的,包括表面形态、多孔结构等等。
4. 临床使用方式(如连续佩戴、使用前溶解等)
根据接触时间,医疗器械可分为短期(<24h)、长期(24h-30d)和持久接触(>30d)器械。按照接触方式和接触组织分类一般分为:不接触人体、表面器械(完好表面、损伤表面、黏膜)、外部接入器械(组织/骨/牙本质、血路,间接、循环血液)和植入器械(骨、组织、血液)。
Endolee,公众号:器械研发那些事
浅谈医疗器械生物学评价的技术评审关注点
接触方式前面也都列的比较清楚了,包括标准里面也都列了这种方式,是不是有连续使用佩戴、累积接触的情况。还有有些产品它是进入人体,它会经过一些处理,比如说那个要进行一些溶解,然后再进行一个注射,这些使用方式进入人体的方式是不一样的,这些都是要进行等同性比较的。
如浅谈医疗器械生物学评价的技术评审关注点介绍所述,只有当以下任何影响生物相容性的因素完全一致时,基本认定符合路径1。这些因素包括:
产品材料化学组成、各组成材料比例、产品物理结构、表面特性、原材料供应商及技术规范、生产工艺、灭菌方法、内包装材料(如适用,主要涉及液体类产品、湿态保存产品)。在申报产品与本企业同类产品存在差异时,需考虑这些因素,并结合相关验证资料进行评价,需要说明的是我们在做生物学试验时选择替代品的依据也是从以上方面一一对比。
需要说明的是因为不同的原材料的供应商,它原材料,不同的原材料,那种本身生物学风险就不一样了,包括像聚乳酸,常见的这种可吸收的这种聚合物材料,它有多种不同的聚合方式,那同样料式聚合方式,溶液聚合,它可能也会用到不同的引发剂。对于液体类的,还有一些湿态保存的料,要考虑它的内包装材料,所以这些如果是完全一样的话,那可以基本认定它是具有一个等同性的。那么这时候如果是自己企业自己申报一个新的产品,它和它的潜在产品-已经上市的产品来进行比较的话,那这时候基本上就是提供一些声明以及证明性文件,比如说供应商一致,这些基本上就不需要再提供其他额外实验资料了,基本上就可以认定它等同性。
如果是和其他家产品对比:你要引起一个等同性判定,这时候可能就需要开展化学表征数据,进行一些可滤沥物的分析,通过这块来判定它的毒理学具有一个等同性。关键原则是:
拟用材料和生产过程引入物质所具有的毒理学或生物相容性不低于同类临床己确定材料(对照市售产品)。
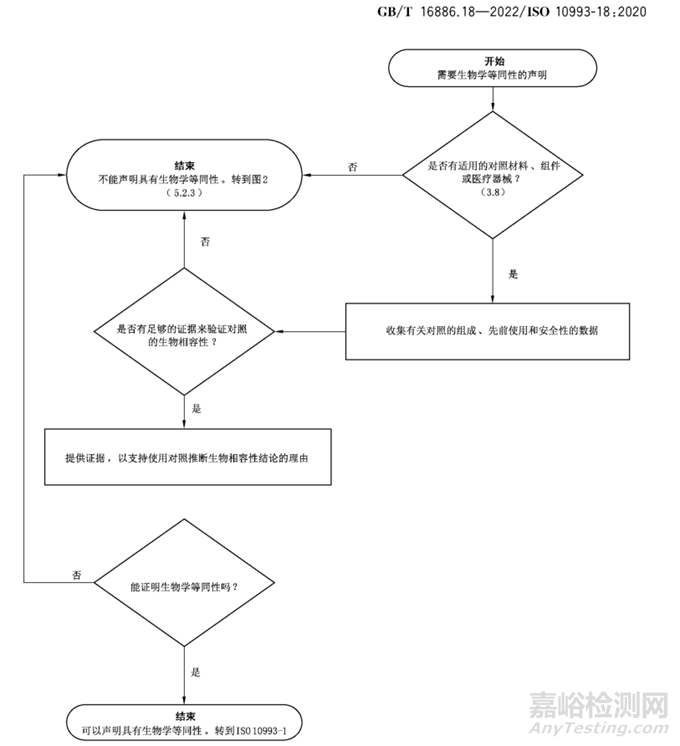
图2 生物学等同性流程图
然后GB/T16886.18的附录c里面它给出来那么8条判定毒理学等同性原则,包括进行颗粒物分析,或者跟一上市产品才是等同的。具体如下:
- 拟用材料的成分和可溶出物与临床己确立材料等同;
- 拟用材料与现行标准中规定的预期用途、接触时间和侵入程度;
- 拟用材料己在比其拟用接触方式侵入横渡更高接触中被确立;
- 拟用材料的可沥滤物限量不超过GB/T 16886.17 -ISO 10993.17规定的允许限量;
- 拟用材料中含有的化学物质或残留物比其所取代的临床己确立材料更具毒理学安全性(假定接触相同);
- 拟用材料中含有的化学物组分或残留物比临床已确立材料中所替代物具有更好的毒理学安全全性(假定接触相同);
- 在可溶出物成分种类与总量不变的情况下,批用材料 临床已确立材料仅有的区别是前者中的添加剂/污染物/残留物水平比后者有所降低或己经排除;
-在相对总量不变的情况下,拟用材料与临床己确立材料仅有的区别是前者所使用的加工条件与后者相比产生的可溶出物水平有所降低。
还有申报产品与现行标准中规定的预期的用途接触方式侵入程度是相当的,然后相关的可滤沥物限量比市售产品要高那么一点,也没有关系,那就评估一下这一种物质高出来的程度是不是在人体可接受的安全阈值之内?如果说还是安全的,那也可以基本上判定它毒理学的等同性。然后具体的这些大家可以下来来看看相应标准,在实际进行毒理学等同性判的时候,看一下实际的产品和市售产品相比,是不是能够建立等同性。
第二部分:路径2-通过可沥滤物化学表征及毒理学分析的方式进行评价
如果说图1绿色线以下有一条是否,那么就得选择路径2进行了。总体来说,路径2是通过可沥滤物化学表征及毒理学分析的方式进行评价。
路径2的第一个框图,风险评定是否有充分论证和/或临床相关数数据,包括化学、生物、物理方面数据。第二个框图是是否有该产品中所有化学物质的充分毒理学的数据,那这块主要强调的是材料是不是已经有了相同应用或者说应用程度,接触人体的水平更苛刻的一种临床的安全使用史,还有相关信息是不是能能够获得很好的展示。所以你说你用的牌号的材料,某某企业它也用过,怎么怎么证明呢?是不是有注册证书上有相应体现,还是说你是通过其他方式获得的那信息需要提供,需要明确。然后这里边提到的所有的化学的物质,就是包括材料和包括了可滤沥物物质的各类的这种材料,尽可能去获取这种所有的材料毒理学数据,比较预期接触剂量和安全剂量,比如说和耐受摄入值,预期产品里边和在人体当中植入量是多少?按表面积算也好,按重量算也好,植入量和安全剂量,Ti是不是低于 Ti,还有一些成分确实是缺乏相应毒理学数值,但是成分是比较恒量的特别少,也可以考虑用提取一些毒理或阈值的方法,这些相关的 GB/T16886系列标准都有具体的介绍。
路径2里边后边的两个框图:我们需要关注数据是否适用于化学混合物,即材料方面。医疗器械所使用的材料种类繁多,机械用材料亦然。此外,在提取这些材料的过程中,以及材料在降解过程中产生的降解产物和衍生物,都可能发生相互作用,导致毒性相较于单一物质时更高或更低。其次,原位聚合的化学反应可能会在人体内产生新的物质,这意味着我们需要开展新的毒理学研究以评估相关风险。最后,关于化学物质的实用性,我们可以一方面查阅文献,另一方面参考类似物质进行分析,以便进行全面评估。总之,在医疗器械领域,毒理学研究至关重要,以确保产品安全可靠。
另一个框图探讨了这些数据是否与接触剂量和途径相关。在毒理学评价中,我们制定了人体可接受的阈值。这些阈值是基于致畸性或全身毒性实验得出的。对于通过不同途径(如经血液注射、吸入呼吸或经口)获得的毒理学数据,需要谨慎外推,并提供充分的依据。如果非要外推,需要确保化学物质的安全阈值不会被超出。
对于路径2,GB/T16886.18有相应的化学表征流程图。其中关键原则是确定所有定量组分,采用浸提法估计临床接触的量,确保每种化学物质的量都不超过毒理学的安全阈值。
这部分内容涉及允许限量建立过程、毒理学值 Ti 的确定以及相关因子的应用。当路径2的框图出现否定情况时,将进入路径3。
第三部分:路径3为通过生物学试验的方式进行评价
关于路径3通过生物学试验方式进行评价,前面撰写过一篇详细的攻略,具体参见呕心沥血篇 | 浅谈医疗器械生物相容性评价。本文就不过多详细的介绍了。
路径3的第一步是根据材料的化学性质和接触类型、时间对器械进行进一步评估。化学性质包括临床使用历史,对于全新材料,需要考虑慢性毒性、致癌性和毒代动力学等方面。如果接触人体后会发生持续反应,可先在体内和体外开展实验,让物质发生反应,再进行生物学实验,这也是一种生物学评价方法。接触类型和时间与生物学终点项目的选择有关。
针对器械的评价针对的是终产品,而非原材料。如确实需要选择替代品,需要说明具体的理由,并对产品材料化学组成、各组成材料比例、产品物理结构、表面特性、原材料供应商及技术规范、生产工艺、灭菌方法、内包装材料一一进行分析,对比差异性,最好做到无差异性。器械在生产加工过程中(包括灭菌过程)可能产生新物质。路径3的后续框图涉及生物学终点的选择、实验及豁免建议的论证。这里提到了常用的表 A.1,用于不同接触类型实验终点选择。需要注意的是,表 A.1 并非实验核查清单,并非所有产品都需要按照清单进行实验选择。它仅作为评价终点,即需要评估生物学终点。但如果已有充分数据,可能无需进行额外生物学实验。
对于特定器械,可能需要额外考虑。尽管这些器械在图中未明确提及,但相关研究仍需开展。例如,非血管支架用于恶性肿瘤治疗时,无需进行毒性研究,因为它们在癌症患者生存期内保留植入产品。表 A.2 中有关终点的说明需基于风险考虑。相较于前一版本的 A.1,表 A.1 涉及的终点显著增多,说明从风险角度出发,需要考虑更多因素。但并非所有风险都需要进行实验,A.1 提供了不同接触性质、组织类型、接触时间的生物学评价终点。其中,物理和/或化学信息是必备的。各个终点多数情况下需进行评估,但鼓励利用现有数据进行评估。全身毒性可通过动物实验研究获得相关信息,无需开展标准化实验。致癌性方面,许多物质可能通过人体某一部位接触产生可浸提物,进入全身循环,因此需考虑潜在致癌物质。此外,要考虑生殖发育毒性,尤其是针对新型材料、已知毒性材料和特定人群的接触生殖器官的器械。最后,提供生物降解信息。
无论哪个路径,最后都需要进行毒理学风险评估,并参考附录 B。附录 B 包括背景信息、生物学评价风险管理行为实施、风险管理指南和特定领域评价指南。可以根据附录 B 撰写生物学评价报告,从而完成生物学评价。