摘要:本文总结抗体药物质量研究的共性问题,包括 国内外法规技术指南介绍、细胞株的质量控制、抗体药物的表征分析、抗体药物的纯度和杂质分析和生物学活性一共五个部分,旨在为从事生物制药研究者提供借鉴。
一、 国内外法规技术指南介绍
近年来医药工业对于生物大分子药品质量控制实践经验的不断积累、分析手段的持续进步,以及质量源于设计、风险评估等国际先进药品质量控制理念的逐步推广,推动了各类生物药质量控制国际指南、法规的修订。抗体类药物属于重组技术产品,质量控制方面应满足国内外重组技术产品相关要求。目前新版《WHO重组DNA产品质量安全有效性评价指南》(WHO Guidelines on Quality, Safety, and Efficacy of Biological Medicinal Products Prepared by Recombinant DNA Technology)已颁布;《欧洲药典》(Monoclonal Antibodies for Human Use)《美国药典》(Control Strategies for RecombinantTherapeutic Monoclonal Antibodies)均设立了针对抗体类产品的总论,即 General Monograph或 General Chapter;ICHQ5、Q6、Q8-Q11等技术指南为抗体类产品的质量控制提供了指导性的范本。由于众多生物药物面临专利保护到期,为了确保生物类似药的质量与安全,欧盟于2006年首先颁布实施了生物类似药的指南,WHO和FDA也先后于200年和2012年颁布了生物类似药指南,并影响了亚洲、南美洲、中东、非洲等医药工业相对欠发达的国家和地区类似法规的制定。近两年这些国家都对已颁布的生物类似药指南进行了修订,WHO也新增了针对单克隆抗体生物类似药的技术指导原则。
中国也于2015年2月发布了《生物类似药研发与评价指导原则》。上述指南和法规中有大量是关于生物大分子量控制技术要求的论述,使包括抗体在内的生物大分子药物质量有了进一步提升的空间。
《中国药典》(三部)颁布了我国单克隆抗体类生物治疗药物的总论和重组DNA技术产品总论,在此基础上制定相应品种各论,对新产品的质量标准化研究具有重要的指导意义。在抗体类药物质量控制方面,应当依据或参照以上药典标准进行,同时也应参照《人用重组DNA制品质量控制技术指导原则》《人用单克隆抗体质量控制技术指导原则》等相关国内法规和技术指导原则的要求。
二、细胞株的质量控制
细胞株的质量控制在《中国药典》(2020版)(三部)通则“生物制品生产检定用动物细胞基质制备及检定规程”中有非常具体的规定,用于生物制品生产和检定的细胞株需通过全面的检定,目前单抗的细胞株主要有鼠源的SP20、NS0和CHO细胞株这几种细胞除一般的细胞株的特征外,还可能携带鼠源性的病毒等外源因子的污染,特别是自身携带的逆转录病毒,动物试验显示具有致肿瘤性,因此必须严格控制细胞株的质量。申报单位不仅要提供规定十分详实的文件资料,还要有具体的试验结果,特别是包括病毒灭活工艺在内的工艺验证资料,并经过国家有关管理部门的认可或批准。
2.1 重组工程细胞的构建
宿主细胞的选择
宿主细胞的选择主要基于两个方面的考虑:安全性及适用性。基于安全的考虑,宿主细胞的来源及培养历史应十分清楚,可溯源有关背景资料,例如,最初分离建立株/系的机构、是否有外源添加序列、传代经过及传代过程中所用过的人源或动物源性材料、细胞可能存在的内源病毒及致瘤性等。对于改良的传代细胞、全新构建的转化细胞等,由于前期研究、背景资料和致瘤性风险等积累的认识十分有限,其开发应用将引人新的安全性隐患,需要慎重权衡利弊,在开展充分研究的基础上严格控制潜在的风险性。
构建过程中的质量控制
目前可采用多种表达载体、基因导入方法和筛选标记等进行工程细胞的构建及筛选,构建成功的标志是工程细胞能够稳定、高效地表达结构正确且具有生物活性的抗体。构建过程应有详细的克隆基因的序列,包括插入的抗体基因及表达载体两侧端制区的核苷酸序列;应详细说明载体引入宿主细胞的方法、载体在宿主细胞内的状态(是否整合到染色体内)及拷贝数;应提供宿主和载体结合后的遗传稳定性资料;应详细叙述在生产过程中,启动和控制克隆基因在宿主细胞中的表达所采用的方法及表达水平。
2.2 细胞库的建立
细胞库的建立是为了保证重组单抗生产的稳定性及批间一致性。参照《中国药典》(2020版)(三部)中“生物制品生产检定用动物细胞基质制备及检定规程”的要求,细胞库为三级管理,即原始细胞库、主细胞库及工作细胞库。如为引进的细胞,可采用主细胞库和工作细胞库组成的二级细胞库管理。在某些特殊情况下,也可使用主细胞库级细胞库,但须得到国务院药品监督管理部门的批准。
原材料的选择及细胞操作要求
用于建库的初始工程细胞株应为经过克隆选择而形成的均一细胞群体,必要时须经与实际生产过程采用的无血清培养基和培养条件相一致的适应性培养。细胞培养及扩增所用原辅料应按照国家药品监督管理总局有关规定执行;动物源性原料的使用应提供来源及质控检测资料;细胞培养液不得含有人血清,不得使用青霉素或β-内酰胺类抗生素;
细胞操作过程应符合现行版《药品生产质量管理规范》的要求。生产人员应定期检查身体。为避免微生物污染和实验室中其他类型细胞的交叉污染,在生产区内不得进行微生物或非生产用细胞的操作;在同一工作日进行细胞操作前,不得操作或接触有感染性的微生物或动物。
原始细胞库(PCB)
重组工程细胞经过克隆培养形成的均一细胞群体,通过检定可用于重组单抗的生产,在特定条件下,将一定数量、成分均一的细胞悬液,定量均匀分装于安瓿,于液氮或-130℃以下冻存,即为原始细胞库,供建立主细胞库用。
主细胞库(MCB)
取原始细胞库细胞,经过一定方式进行传代、增殖后均匀混合成一批,定量分装保存于液氮或-130℃以下,经全面检定合格后,即为主细胞库,用于工作细胞的制备。
工作细胞库(WCB)
工作细胞库的细胞由MCB细胞传代扩增制成。由MCB的细胞经传代扩增,达到定代次水平的细胞,合并成一批均质细胞悬液,定量分装于安瓿或适宜的细胞冻存管,保存于液氮或-130以下备用,即是工作细胞库。生产企业的工作细胞库必须限定为一个细胞代次,冻存细胞的传代水平需确保细胞复苏后传代增殖的细胞数量能满足生产一批制品。复苏后细胞的传代水平不应超过批准用于生产的最高限定代次。
2.3 细胞库的管理
细胞库中每支细胞应有明确的标记,包括细胞系/株名、代次、批号、编号、冻存日期、储存容器的编号等;冻存前的活细胞比例应不低于90%,复苏后细胞存活率应不低于80%,冻存后的细胞应至少做一次复苏培养并连续传代至衰老期,检查不同传代水平的细胞生长情况;主细胞库和工作细胞库应分别存放,非生产用细胞应与生产用细胞严格分开存放。上述各级种子库的细胞应按照特定的要求经过全面检定合格后方可使用。
2.4 细胞的检定
合格的生产细胞是得到合格的重组抗体的前提和基础,为保证产品的安全性和有效性,应对生产细胞进行全面的检定。《中国药典》(2015版)(三部)中“生物制品生产检定用动物细胞基质制备及检定规程”及“重组制品生产用哺乳动物细胞质量控制技术评价一般原则”对细胞的质控内容作了较为详细的阐述,主要包括细胞的鉴定及微生物污染的检定。重组单抗生产细胞作为经过DNA重组技术获得的含有特定基因序列的细胞系,在质控内容方面还增加了细胞稳定性的考察、抗体基因或抗体的鉴别试验、细胞产物中外源病毒因子的检测。
由于生产细胞分三级细胞库管理,对于原始库细胞和/或主库细胞,通常需要进行一次全面系统的研究检定,包括遗传学、生物学和微生物学检定,以保证起始细胞的一致性并排除污染。经过传代稳定性研究的主细胞到工作细胞,只经过简单的传代、扩增,可适当简化检定项目,重点检测外源因子污染和细胞污染。
细胞的鉴定
细胞鉴别试验:现行药典规定,新建细胞库(MCB和wCB)及生产终末细胞应进行细胞鉴别试验,以对细胞的种属来源进行确证并排除其他细胞的交叉污染。细胞鉴别可通过生长形态、生化检测法(如同工酶法)免疫学检测(如组织相容性抗原、中和特异性免疫血清)遗传学检测(如染色体核型、标记染色体检测)遗传标志检测(如DNA指纹图谱、STR图谱、基因组二核苷重复序列)等方法进行;
抗体基因及抗体的鉴别试验:抗体基因的检测是抗体生产细胞鉴别试验的重要组成部分,其目的是保证生产细胞中抗体基因的正确与完整,以得到结构正确的抗体产品。常用的方法包括:通过PCR扩增样本DNA或用从细胞基质中分离的RNA制备的cDNA来进行DNA序列分析;通过限制酶酶谱和 Southern杂交来检测基因的完整性确定目的基因的拷贝数,并检测是否有任何序列插入或缺失等。目的基因的检测应贯穿于工程细胞的构建及筛选、细胞库的建立和检定,以及生产细胞培养监控的全过程。抗体产物可通过免疫斑点或免疫印迹试验等进行鉴别;
致瘤性鉴定:目前生产重组单抗经常用到的细胞株CHO、NS0、C127等已证明具有致瘤性,国外指导原则通常不再要求进行致瘤性检查,但对于该细胞来源的制品,要优化生产工艺,严格限制致瘤性成分如宿主残余DNA在成品中的含量;现行药典也规定对于已证明具有致瘤性的传代细胞可不必进行致,性检查,但一般认为生产中应用的插入外源基因的工程细胞应当视为全新的细胞,必须进行致瘤性的检查,并在细库检定中进行传代/扩增过程中的对比试验,观察致瘤性特征的改变,并制定后续处理工艺条件和限制性要求,对致瘤性带来的安全性风险进行控制;
致瘤性试验可通过体内试验和体外试验进行。体内试验有裸鼠法和新生小鼠法,体外法可采用软琼脂克隆形成试验或器官培养试验,具体实验操作参见现行药典中“生物制品生产检定用动物细胞基质制备及检定规。
病原微生物检测
细菌、真菌及支原体检查:对细胞培养上清液进行无菌检查,结果须符合规定要求。使用的培养基应适合需氧菌、厌氧菌和真菌的生长,并且培养基的灵敏度要满足要求。无菌检查法包括直接接种法和薄膜过滤法,如供试品允许,应优先采用薄膜过滤法。对于直接接种法,若制品中含有防腐剂,应先行增菌培养。在进行支原体检查时,应注意同时进行培养法和指示细胞法两种方法;
内、外源病毒因子检查:用于确定待检细胞中是否存在潜在的可感染的病毒如鼠源性病毒)及操作带人的外源性病毒。检测病毒的种类及方法须根据细胞的种属来源及细胞特性决定,对于重组工程细胞来说,还应当对细胞裂解物或收获液进行外源因子检测。
一般病毒因子可通过细胞形态观察及红细胞吸附试验、不同细胞传代培养法及接种动物和鸡胚法检测,其中《中国药典》(2015版)(三部)中新增规定:用不同细胞传代培养法检测病毒因子时,应设立病毒阳性对照,包括可观察细胞病变的病毒阳性对照及血吸附阳性对照。另外,新版药典对检定用细胞也增加了明确要求,具体参见《中国药典》(2015版)中“生物制品生产检定用动物细胞基质制备及检定规程”。对于逆转录病毒及其他内源性病毒或病毒核酸的检测,现行药典规定:小鼠来源和其他啮齿类来源的细胞系或其杂交瘤细胞系有可能携带潜在的逆转录病毒。因此,对于人-鼠杂交瘤细胞系应进行特异性逆转录病毒检测。如用于单克隆抗体生产的小鼠细胞系,则可不检测特异性的逆转录病毒,但在生产工艺中应增加病毒灭活程序。根据待检细胞系/株的种属及组织来源,还应进行特殊外源病毒因子的检测。如为鼠源细胞系,一般检测出血热病毒、淋巴细胞脉络从脑膜炎病毒、Ⅲ型呼肠孤病毒、仙台病毒、脱脚病病毒、小鼠腺病毒、小鼠肺炎病毒、逆转录病毒等;如为人源细胞系,应检测人鼻咽癌病毒、人巨细胞病毒、人逆转录病型肝炎病毒、人丙型肝炎病毒等。
细胞株的稳定性研究
抗体生产细胞一般为通过基因工程获得的重组工程细胞,生产者须具有相关的稳定性研究资料,包括重组细胞的遗传稳定性、目的基因表达稳定性、目的产品持续生产的稳定性,以及一定条件下保存时细胞生产抗体产品能力的稳定性等资料。
三、抗体药物的表征分析
单克隆抗体是迄今为止分子量仅次于重组人凝血因子Ⅷ的药用蛋白,其分子结构具备高度的复杂性。IgG型抗体是由两条重链(≈50kDa)和两条轻链(≈25kDa)经链间二硫键相连,所构成的“Y”形四聚体糖蛋白。一般在抗体的恒定区或可变区还存在MO糖基化修饰。抗体药物与药物靶标特异性结合后,通过阻断细胞信号转导通路,或诱导效应功能,如抗体依赖的细胞介导的细胞毒性作用(ADCC)补体依赖的细胞毒作用(CDC)、抗体依赖的细胞介导的细胞吞噬作用(ADCP)或运载偶联小分子化药等多种方式发挥作用。
由于重组抗体的制备工艺采用动物细胞异源表达,其分子结构上存在多种翻译后修饰。因此,抗体药物的结构还具有显著的“异质性”或“非均一性”的特点。根据IgG型抗体潜在的修饰位点(NO糖基化、焦谷氨酸环化、赖氨酸剪切、天冬氨酸异构及甲硫氨酸氧化等),推测出的理论变异体至少有108种,这些修饰变异又表现为分子大小、电荷、糖谱等多种形式的差异。临床研究已经证实,某些抗体药物的变异体具有不同的药代、药效和免疫原性属性。可以说,抗体分子结构上存在的“复杂性”和“异质性”,决定了抗体药物表征研究的难度与挑战。
正确的分子结构是保证抗体药物安全、有效的物质基础。质量研究中对抗体药物进行全面的表征研究是进行质量控制的重要手段。因此,国内外关于抗体药物研发的指导原则均强调,抗体药物的表征研究应采用现有先进的分析手段( state-of-the-art),从物理化学、免疫学、生物学等角度对产品进行全面的分析,并提供尽可能详尽的信息以反映目标产品内在的质量属性。下文将结合抗体药物具体案例,介绍抗体药物表征研究的主要内容与一般方法。
一级结构
分子量
由于抗体的恒定区具有糖基化修饰,一般经过脱糖处理后测定分子量。通过实测值和理论值的比较,可以初步判定抗体药物序列是否正确。例如,某曲妥珠生物类似药完整蛋白分子量与原研参比存在64Da差异,重链分子量与原研参比存在32Da差异,因此,可初步判定该生物类似药重链氨基酸序列存在错误或翻译后修饰。
氨基酸序列
抗体药物的一级结构(氨基酸序列)是其生物活性、临床疗效的物质基础,尤其是对于生物类似药而言,确保氨基酸序列与原研参比一致是首要条件。一般重组抗体经蛋白酶切(LsC、AspN或GuC)后,利用串联液相质谱技术进行肽质量图谱分析来确证氨基酸序列。例如,上文提及的曲妥珠生物类似药经肽质量图谱分析确定,其分子量差异位于胰蛋白酶酶切肽段T35上。在生物类似药开发的早期筛选阶段也会出现一级结构的改变,均可以利用质谱技术予以鉴定。
氨基酸含量分析
重组抗体酸水解后,经衍生化试剂处理后可使用液相定量分析其氨基酸含量。但是由于水解过程中对于稳定性差的氨基酸(丝氨酸、苏氨酸等)破坏较大,或某些氨基酸因空间位阻原因难于水解(异亮氨酸),某些氨基酸的实际测定值偏低。此外,对于生物大分子而言,氨基酸含量仅是其一级序列确证的佐证,并不能表明其氨基酸序列的正确性。因此,氨基酸含量分析在抗体药物的结构确证研究中意义有限。
N/C端异质性
N端测序是抗体药物一级结构鉴定的重要方法,通常将还原后的抗体轻、重链经Edman降解依次测定N端氨基酸序列。若抗体的N端存在焦谷氨酰封闭,需采用焦谷氨肽酶去封闭后再进行 Edman降解测定。重组表达的抗体药物由于工程细胞内羧肽酶D的降解,还会导致重链C端赖氨酸不完全剪切。目前,已经证实重组抗体普遍存在着NC端异质性的现象,并且没有证据表明NC端异质性对抗体的安全性、有效性产生影响。但是,通过NC异质性的分析有助于加强重组抗体药物的质量控制。为了减少C端的异质性,亦有通过基因水平去除末端赖氨酸的设计。
氨基酸修饰分析
抗体药物的质量肽图通过测定肽段分子量及二级碎片分子量,可以进一步分析氨基酸的修饰类型及其比例,如脱酰胺、甲硫氨酸氧化、糖基化修饰、N端焦谷氨酸环化、C端赖氨酸切除等。目前已经证实在加速降解条件下,抗体CDR区氨基酸的化学修饰可能会影响其亲和力和生物学活性。鉴定修饰的类型及位置可作为抗体药物CQA评定的重要指标。
糖基化修饰
除了个别改构的IgG1(N297A)型抗体不具有糖基化修饰外,绝大多数抗体和抗本融合蛋白均存在M糖或O-糖修饰。抗体的M糖修饰发生在“Asn- X-Ser/Thr”(X为除Pro外的任意氨基酸)序列中的Asn位点。O糖修饰没有特殊基序结构,多发生在抗体融合蛋白序列的Ser和Thr的羟基上。糖基化修饰在维持抗体正常结构和生物活性上发挥着重要作用。例如,高半乳糖修饰可提高抗体的CDC效应;低岩藻糖修饰可提高抗体的ADCC、ADCP效应;而a1,3半乳糖或NGNA等非人糖基化修饰则可在临床上引起免疫原性等,
因此,需要对糖基化位点、寡糖分布及糖链结构等进行充分研究。例如,阿巴西普存在3个M糖基化修饰位点和4个O糖基化修饰位点;依那西普含有3个N糖修饰位点和13个0糖修饰位点;西妥昔单抗在Fab和Fe区域均具有M糖修饰。寡糖分布是对抗体药物中不同修饰形式的寡糖链所占比例进行分析。一般使用糖苷酶酶切糖链后,经衍生化处理,使用液相色谱分析确定各寡糖链所占的比例。在抗体药物的质量控制中,对于影响抗体效应功能的寡糖分布,应重点关注并进行控制。例如,以ADCC效应为主要作用机制的曲妥珠单抗,应重点关注影响恒定区效应功能的寡糖分布,但是曲妥珠单抗原研产品不同批次的糖基化修饰亦有较大的变异。糖链的结构分析一般釆用多种糖苷酶分步酶切确定单糖连接方式,通过测定糖链分子量预测糖链结构。
二级结构
二硫键
IgG型抗体由两条轻链和重链,通过链间二硫键组装而成。二硫键的构型有时可以显著影响抗体的功能。二硫键的测定通常采用酶切后质量肽图谱的方法,通过测定还原与非还原状态的酶切分子量或通过二硫键肽段的二级质谱碎片来确证抗体二硫键配对情况。抗体药物的表征研究既要验证正确的二硫键配对,也要关注二硫键错配形式。对于治疗性抗体应用较多的IgG亚型:IgG1和IgG4抗体分子内含有16个二硫键,其中链内二硫键12个,链间二硫键4个;1gG2抗体分子内含有18个二硫键,其中链内二硫键12个,链间二硫键6个;而对于复杂的抗体融合蛋白依那西普,则含有29个二硫键和多种错配形式二硫键。
自由巯基
理论上含完整二硫键的IgG抗体分子内应不存在自由巯基,但是由于有些抗体存在额外的半胱氨酸或者存在未形成二硫键的半胱氨酸残基,抗体分子中一般含有少量的自由巯基(0.02 mol/mol蛋白左右〉通常采用 ELLMAN试剂法测定自由巯基,其原理为DTNB与抗体的自由巯基反应后生成TNB,根据TNB吸光度确定抗体的自由巯基含量。
高级结构
测定抗体分子高级结构的常用方法是圆二色谱法,该方法利用蛋白质的圆二色性及不对称分子对左右圆偏振光吸收的不同来进行结构分析。远紫外区(190-230mm)光谱可反映蛋白质二级结构,即α螺旋、β折叠、转角和不规则卷曲的比例。近紫外区(250-350mm)光谱反映蛋白三级结构变化,即侧链生色基团色氨酸、苯丙氨酸、酪氨酸等残基的排布信息和二硫键微环境的变化;此外,差示扫描量热法、氢氘交换质谱傅里叶转换红外光谱X光晶体学核磁共振技术等也常用于分析抗体药物的高级结构。
免疫学活性
可变区抗原亲和力
抗体药物的靶向特异性体现在其可变区与靶抗原特异性结合,可变区的抗原亲和力一般采用酶联免疫吸附法、流式细胞术和BA法等方法进行测定。采用竞争ELSA法测定抗体结合活性时,将供试品与标准品与酶标抗体竟争结合包被抗原,对抗体浓度与吸光值的量效关系进行四参数拟合后,计算供试品与标准品的ECs值来评价抗体药物的结合活性;流式细胞术通过测定抗体与表达抗原的靶细胞之间的结合阳性率,评价抗体与细胞表面抗原的结合活性;BIA法是通过实时、动态监测抗体与芯片表面固化的抗原之间的结合解离反应,计算出抗体的亲和常数等动力学参数。
恒定区效应功能
抗体的恒定区通过与FcR结合介导ADCC、ADCP等功能,通过与C1q结合介导CDC功能,通过与新生儿受体FRn结合延长抗体在体内的半衰期。因此,可以通过测定抗体与相应受体结合力(CD16,C1q)来间接反映其潜在体内效应功能,也可以采用基于细胞的生物测活方法直接测定恒定区介导的效应功能。例如,采用新鲜分离的人外周血单个核细胞或NK细胞作为效应细胞,或者采用报告基因法测定抗体介导的ADCC效应;采用补体与靶细胞共孵育的方法,测定抗体介导的CDC效应。
生物学活性
抗体药物的生物活性直接反映了其临床应用的体内效力,是抗体药物质量控制的重要指标。抗体药物的生物学活性测定,一般采用体外细胞法或动物模型法模拟药物的体内作用机制,并通过与活性标准品的比较对其量效关系进行赋值评价。
常用生物活性测定方法主要有细胞增殖抑制法、细胞毒性法、抗体依赖性细胞介导的细胞毒性法和补体依赖的细胞毒性法等。例如,靶向细胞生长因子(VEGF、Her2、BGFR等)抗肿瘤单抗均采用表达靶抗原的细胞( HUVEC、BT474、DiFi等)进行细胞增殖抑制实验进行生物测活;靶向TNF-α的抗体或融合蛋白,根据拮抗TNF-α对敏感细胞系(L929细胞)的杀伤活性测定生物活性;利妥昔单抗通过测定介导补体对靶细胞的杀伤测定生物活性等。
近年来,除了上述采用原代细胞生物测活外,通过导入抗体靶标和报告基因构建的转基因指示细胞株开始应用于抗体药物的生物测活。例如,靶向PD-1或PDL1单抗采用稳转NFAT报告基因和人PD1的 Jurkat细胞作为效应细胞,采用高表达人PDL1的CHO细胞作为靶细胞。在效应细胞与靶细胞共孵育条件下加入抗体,根据荧光素酶检测系统评价抗PDL1抗体生物活性。
对于抗体药物而言,表征研究始终贯穿着药物研发的生命周期。申请临床前,在工艺开发、CQA、CPP评估、质量研究和稳定性研究中对候选药物进行充分的表征研究有助于尽早锁定生产工艺,确定批次放行质量标准和产品的储存条件及有效期;产品申请上市前,对工艺验证批次产品的质量进行表征研究,可以证明拟上市规模下生产工艺的稳健性和产品质量的一致性;在产品上市后,如果发生生产工艺的变更,要提供变更前后多批次产品的表征研究数据,以支持工艺变更前后产品质量具有“可比性”。此外,近年来国内外正在兴起抗体生物类似药研发的热潮。其中,对多批次原研参比和生物类似药进行全面表征比对研究,也是证实生物类药具备“质量相似性”,进而减免非临床、临床研究的关键所在。
四、抗体药物的纯度和杂质分析
4.1 大小异质性分析
纯度测定是重组蛋白药物的一项重要检测指标,该指标直接反映了抗体纯化工艺水平及产品质量的高低。抗体纯度测定主要涉及两个方面的问题。一是抗体分子与杂质的有效分离。在实际测定中,某些产品相关杂质,如肽链截短或延长形式、修饰形式、聚合体、多聚体等,由于性质与主蛋白比较接近,分离起来可能有些困难,须选用合适的分离方法。二是对主蛋白及杂质的检测与定量。常用的检测器有紫外检测器、凝胶成像扫描仪、荧光检测器、电化学检测器、示差检测器、蒸发光检测器、质谱检测器等。由于各种检测器灵敏度的限制,有些生产纯化过程及操作环境中引人的微量杂质不易检测到。为避免一种检测方法在蛋白纯度检测中的偏差,一般选用至少两种分离方法进行检测,以得到相对准确的纯度信息。测定抗体纯度的常用方法是非还原型或还原型SDSPAGE或 CE-SDS法、分子排阻色谱法( SEC-HPLC)等方法,对单体、聚合体或片段进行定量分析,如供试品具有Fc效应子功能,则还需关注非糖基化重链的情况。供试品测定结果应在规定的范围内。
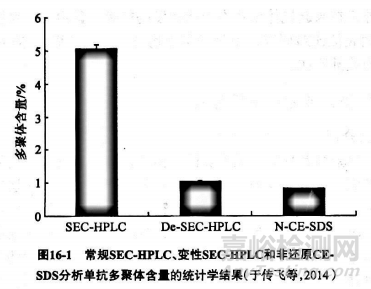
单克隆抗体(单抗)由两条重链和两条轻链构成,相对分子量高达15万Da左右,结构复杂,其中大小异质性是单抗的重要质控指标之一。大小异质性一般可分为三类,即单体、片段和多聚体。片段包括降解的单抗和组装不完全的重轻链等;而多聚体则包括二聚体、寡聚体或更复杂的聚体等,多聚体不仅可能是单抗中的无效成分,还是导致单抗免疫原性的重要原因之一,所以大小异质性是单抗生产工艺优化、生产过程控制及放行分析中不可或缺的检测项目,也是单抗稳定性评价的重要指标之一。随着技术的进步,单抗大小异质性的分析手段不断涌现,不同的分析技术其分析原理不同,所获得的结果反映单抗在大小异质性方面不同的特性。由于 SEC-HPLC及 CE-SDS相对简单易行,成为分析单抗类制品大小异质性的两种常规方法。有研究利用非还原(SDS和碘乙酰胺处理)及常规(无处理)和变性(SDS和碘乙酰胺处理) SEC-HPLC评价了一种抗ⅤEGF单抗的大小异质性,初步阐明了二者结果差异的原因,为单抗制品的质量控制研究提供了理论基础。
三种分析的结果比较如图16-1所示,可以看出非还原 CE-SDS的多聚体含量分析结果显著低于常规 SEC-HPLO,但与变性 SEC-HPLC的分析结果基本一致。多聚体为共价和非共价结合两种形式总和,而在 CE-SDS分析的情况下,由于去垢剂SDs的存在,非共价形式的多聚体被解离成为单体,所以多聚体的含量相比常规 SEC-HPLC明显降低。在变性 SEC-HPLC分析的情况下,由于SDS的存在,多聚体含量则与非还原CE-SDS的分析结果基本一致。所以与非还原 CE-SDS相比, SEC-HPLC可更加客观地评价样品中总多聚体的百分比含量。该研究分析的单抗样品多聚体主要是以非共价键的形式连接,所以应注意两种方法所得到的多聚体含量结果具有不同的含义,在分析多聚体含量时,常规 SEC-HPLC分析比 CE-SDS分析更加客观。
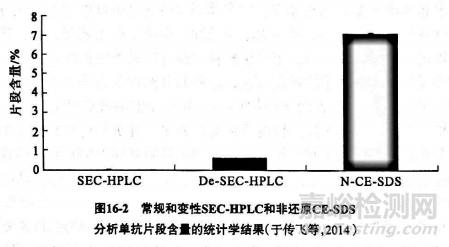
对于片段分析,由图16-2可以看出,非还原 CE-SDS所分析的片段比例显著大于常规和变性 SEC-HPLC,其主要原因是 SEC-HPLO的分辨率较低,无法对单体及大部分片段进行有效分离。单抗分子中往往有少部分重链和/或轻链的链间二硫键连接不完全,以非共价键与单抗其他部分相连接。而SDS可打开非共价键,所以在非还原CESDS分析结果中,除HHLL(全抗分子)外,还可形成HHL、H、H、H、L,以及其他与单抗非共价结合的降解片段;常规 SEC-HPLO由于不能有效解离非共价结合的片段,加之其低分辨率,对片段含量的检测有一定的局限性,所以非还原 CE-SDS和SEC-HPLC二者片段峰所代表的形式并不相同。
上述研究初步阐明了单抗类制品大小异质性的两种常规放行分析方法即 SEC-HPLO和非还原 CE-SDS二者结果差异的原因,为单抗制品的质量控制研究提供了理论基础,为单抗分子大小变异体质检策略提供了技术支撑。
4.2 电荷异质性分析
单抗类制品的多种翻译后修饰可导致其电荷异质性,而某些电荷异质性由于对单抗稳定性及其生物学功能发挥具有重要的影响而成为关键质量属性(CQA),并且电荷异质性可反映其生产工艺的稳定性,所以受到生物技术产业界及监管机构密切关注。成像毛细管等电聚焦电泳是单抗电荷异质性分析的常用分析技术,可利用此方法对单抗电荷异质性峰的组成进行初步研究。
将单抗 Ustekinumab(由SP2/0细胞表达)用羧肽酶B去除C性峰为C端赖氨酸不均一性所引起,如图16-3所示。但是经羧肽酶B酶切后,其主峰前仍有2个比例较高的酸性峰。为判断唾液酸修饰对其电荷异质性的贡献,将羧肽酶B酶切后的抗体继续用M糖苷酶酶切,分析其电荷异质性,结果显示经过2个酶切处理后抗体2的图谱基本上只剩1个主峰,证明抗体2的电荷异质性主要是由C端赖氨酸的不均一性和唾液酸修饰所引起。
由于单抗制品高度复杂的异质性其质控需要组合多种理化分析技术对其进行检测如cIEF、 IEX-HPLC、 ICIEF、疏水高效液相色谱( HIC-HPLO)反相高效液相色谱(RP-HPLC)等方法,尽可能对不同电荷变异体组分进行鉴别,并规定相应的可接受标准。成像毛细管等电聚焦电泳为对其进行分析提供了一个快速、灵敏、高分离度的选择,对保证单抗类生物技术药物生产工艺的稳定性及控制其质量和提高质量标准均具有重要意义。
4.2 杂质分析
采用适宜的方法对供试品氧化产物、脱酰胺产物或其他结构不完整分子进行定量分析。采用适宜的方法对供试品宿主蛋白质、宿主细胞和载体DNA、蛋白A及其他工艺相关杂质进行检测。由于单克隆抗体人用剂量较大,基于方法学灵敏度考虑,目前残余DNA检测常用方法是定量PCR法,样品需要经过前期抽提处理,因此应对抽提及定量PCR整个过程进行方法学验证。重组抗体多由哺乳动物细胞表达产生,在抗体的纯化过程中不可能将宿主细胞蛋白完全去除,残留的异体蛋白进入人体有可能引发免疫反应,故需对残留蛋白量进行限制。宿主细胞蛋白残留量检测中,除杂交法(WB、2DWB、2 D-DIGE)和傅里叶转换红外光谱法外,ELSA方法是目前最常用的方法,其灵敏度高于上述两种方法。
ELISA方法检测HCP效率受所使用抗体对残留HCP的覆盖率的影响,而残留FCP种类受生产细胞株、细胞培养工艺和下游纯化工艺等多种因素的影响。因此需要对商用试剂盒的适用性进行方法学验证,并建议开发工艺相关的HCP检测方法。在蛋白A残留量检测中,对抗体进行纯化时会应用蛋白A亲和柱,柱子型号不同,所用蛋白A也有差异,USP制备了包括原料在内的4种不同类型的蛋白A标准物质:Natural Protein A46800, Recombinant Protein A44600Cys-rpA3430, Mahselec SuRe26700。标准物质的建立对蛋白A含量测定方法的标准化很有意义。蛋白A测定方法多采用ELSA法:将蛋白A的特异性抗体包被96孔板,加入待测样品及蛋白A标准溶液,溶液中的蛋白A与包被抗体结合,洗涤后加入酶标抗体,加底物显色并测定吸光值,通过标准曲线法测定待测抗体溶液中的蛋白A含量。
五、生物学活性
活性测定是对抗体类药物的有效成分和含量及药物效价的测定,是确保药物有效性的重要质控指标。抗体类药物可通过其Fab段结合抗原发挥生物学效应,如大多数抗细胞因子类抗体药物即是通过和细胞因子或其受体结合阻断从而阻断信号通路传导;也可通过Fc段发挥作用,单抗药物与靶细胞表面分子结合后,可介导补体clq结合到抗体的Fe段发挥补体依赖的细胞毒效应( complement dependent cytotoxicity,cDC),重组抗CD20、CD52单抗即是通过测定免疫复合物活化补体产生细胞毒作用后B淋巴蜜细胞的死亡评价其生物学活性;Fc段也可以通过和不同的FeR结合介导免疫效应抗体依赖性细胞介导的细胞毒作用( antibody-dependent cell-mediated cytotoxicity,ADCC)和抗体依赖性细胞介导的吞噬作用( antibody-dependent cell-mediated phagocytosisADCP),如图164所示。ADCC和ADCP是许多针对肿蜜及自身免疫病的抗体类药物重要作用机制之一。
药物的生物学活性测定主要是在体外建立相应的细胞评价模型,模拟其作客观的全程量效反应,并通过与活性标准品的比较对其生物学活性进行评价。近年来,转基因细胞技术和一些新技术也被应用于抗体类药物的生物学活性测定下文将对应用于抗体药物活性评价的传统方法和前沿技术进行简要介绍,为新型抗体药物活性方法的建立提供新的思路。
5.1 基于细胞的生物学活性测定方法
随着药物高通量筛选平台的建立和生产规模的扩大,以及对“3R”( reduction,refinement, replacement)原则理解的不断深化,人们越来越多地寻求动物试验替代方法。而基于细胞系的体外生物活性分析方法,由于其高通量、高效率、高精确度等优势,越来越受到研究者和生产企业的青睐。单克隆抗体药物的作用靶点分为细胞因子及其受体、肿瘤细胞表面抗原、CD分子、病原微生物及其产物、其他靶点。根据抗体药物作用的特点,目前主要有以下几类基于细胞的测活方法。
细胞增殖抑制法
针对生长因子靶点的抗体药物多是釆用细胞增殖抑制的方法来反映抗体的生物学活性,包括抗血管内皮生长因子单抗、抗人表皮生长因子受体2单抗及抗人表皮生长因子受体单抗等。其中,抗VEGF单抗的经典测活方法是人脐静脉内皮细胞增殖抑制法,即在刺激因子EGF存在的情况下,抗ⅤEGF单抗能够以剂量依赖性的方式抑制HVEC细胞的增殖。抗HER2单抗的活性测定通常选取HER2阳性的乳腺癌细胞,如BT474、SK-BR-3、SKOV3、MCF-7等作为靶细胞,抗体与靶细胞表面HER2抗原结合后,能够有效抑制细胞生长信号传递,从而抑制细胞增殖。抗EGFR靶点单抗也是通过特异结合并封闭表皮生长因子受体,有效抑制肿瘤细胞生长,如DFi细胞、A431细胞增殖抑制法等。
细胞毒性法
细胞凋亡有两条途径,一是线粒体依赖途径,另一个为死亡受体介导途径。肿瘤坏死因子a( tumor necrosis factor-alpha,TNF-a)和受体结合后,可启动死亡受体介导途径,使 procaspe8自我水解、活化,形成活性 caspase-8,后者再激活 caspase3、6、7等引起下面的级联反应,导致细胞发生凋亡。重组人Ⅱ型肿瘤坏死因子受体·抗体融合蛋白的活性测定可以利用其能够抑制TNF-a所引起的敏感细胞系U937中 caspase3/活化,通过 Caspase-Glo3/7检测试剂盒中荧光素酶发光信号的改变来反映该制品的活性此外,针对TNF-a的单抗还可以采用对TNF杀伤敏感的细胞系,如小鼠成纤维细胞L929、小鼠纤维肉瘤细胞WEH64等,抗体能够抑制TNF-a所诱导的细胞凋亡作用通过检测细胞存活的染色来评价抗体的生物学活性。
补体依赖的细胞毒法
单抗药物与靶细胞表面分子结合后,可介导补体C1q结合到抗体的Fc段,并于肿瘤细胞膜上形成类似于穿孔素效应的攻膜复合体( membrane attack complex,MAC)造成细胞外离子大量内流,最终导致肿蜜细胞的溶解。CDC生物学活性测定中一个关键环节是补体的选择及对其质量的控制,补体组分多,且热不稳定,容易失活,目前所用的补体来源包括正常人血清、豚鼠、家兔等,来源复杂、剂型不同,因此在CDC实验中需要考虑补体效力、稳定性,尽量减少活性测定反应终点和测定结果的变异。以CD( cluster of differentiation)分子为靶点的单抗药物,如抗CD20单抗、抗CD52单抗等,其生物学活性评价方法主要为CDC活性测定,即将重组抗体进行系列稀释后与高表达相应CD抗原的靶细胞结合,在补体存在的情况下,抗体与细胞表面抗原形成抗原-抗体复合物,激活补体经典活化途径,完成攻膜复合物的装配并在细胞表面打孔最终导致细胞溶解。
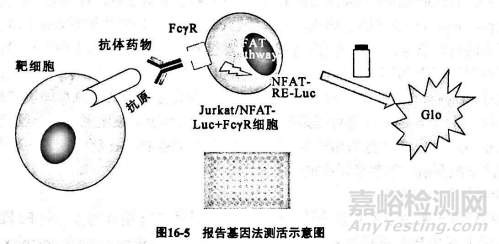
ADCC和ADCP
单抗药物通过Fc段介导的抗体依赖性细胞介导的细胞毒作用(ADCC)和抗体依赖性细胞介导的吞噬作用(ADCP)的传统检测方法多为基于新鲜制备的外周血单个核细胞或者自然杀伤细胞作为效应细胞的杀伤试验,但以上方法存在细胞分离和培养困难、变异大、操作繁琐、高背景值等缺陷。近年来基于转基因细胞法已建立了稳定而可靠的ADCC和ADCP评价方法。ADCC和ADCP报告基因法均使用工程改造的 Jurkat细胞作为效应细胞,分别稳定表达了其效应介导的主要受体FcRl和 CyrIlla,以及由NFAT应答元件驱动表达的荧光素酶报告基因。当高表达抗原的靶细胞与表达FRa或FcRa的 Jurkat细胞通过单抗桥联时,可引起NFAT荧光素酶报告基因的活化,通过检测荧光素酶化学发光信号来反映抗体的ADCC或ADCP效应(图16-5)。该方法操作简便易行,专属性强、重复性好、准确性髙,通过选择不同的靶细胞可作为抗CD20单抗、抗HER2单抗、抗EGFR单抗ADCC生物学活性的常规检查方法,用于评价包括人鼠嵌合单抗、人源化单抗、全人源单抗和糖基化改造单抗在内的各类抗CD20单抗、抗HER2单抗、抗EGFR单抗的ADCC活性;更重要的是,该方法可用于评价糖基化修饰与单抗Fc效应功能的关系,这也为该类制品工艺稳定性评价及结构与功能关系评价奠定基础。
5.2 转基因细胞生物学活性测定方法
转基因细胞法为很多没有强反应性细胞系,或者没有易检测的细胞学效应的生物技术药物活性测定提供了选择。转基因细胞法具有的实验周期短、批间差异小等优势使其更能满足药物批间一致性和稳定性的测定以及监管的需要,因此,转基因细胞法逐渐成为生物技术药物活性测定的趋势,一些传统的生物学活性方法逐渐被转基因细胞法所替代。构建转基因细胞生物学活性测定法首先应该全面深入地研究药物的作用机制,包括受体激活、信号转导、信号传递及终效应,然后,选择合适的靶标作为药物活性测定的指标。目前国内在建立转基因细胞法测定生物治疗药物生物学活性领域已走在世界的前列。中国食品药品检定研究院率先建立了干扰素测活的荧光素酶报告基因法,克服了国际通用的病毒抑制法中由操作活病毒导致的缺点,使检测周期缩短至原来的1/3,并最终纳入《中国药典》(2015版)三部。将这一理念应用于抗体药物生物学活性的测定同样取得了较好的成果,不仅实现了传统测活方法改进的突破,还解决了免疫检查点等一类新药生物活性测定的难题。