对于已在美国FDA列名上市的医疗器械,绝大多数器械都是需要满足FDA器械GMP(即21 CFR 820,通常称之为QSR 820)要求。只不过与其它国家法规不一样,FDA产品上市前,绝大部分器械是无需GMP核查,而是上市后GMP核查,但PMA产品例外,PMA产品(如三类器械)在获批前FDA需要实施GMP现场核查。
一、PMA体系核查法规依据
PMA体考的法规依据和其它器械产品一样,核心为:
21 CFR Part 820 Quality System Regulation
21 CFR Part 803 Medical Device Reporting
21 CFR Part 806 Medical Devices; Reports Of Corrections And Removals
21 CFR Part 821 Medical Device Tracking Requirements(仅限于植入超过1年的器械、生命支持器械,或器械失效时可造成严重不良后果的器械)
另外包括一份指南性文件:1999年8月发布的“质量体系检验技术”(QSIT:Quality System Inspection Technique),为FDA审核员提供检查流程指导,实际审核时,审核员基本按QSIT流程实施审核,因而该份指南非常重要。
二、PMA体系核查要点
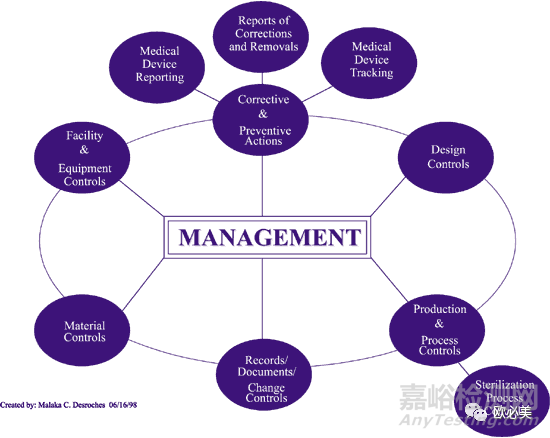
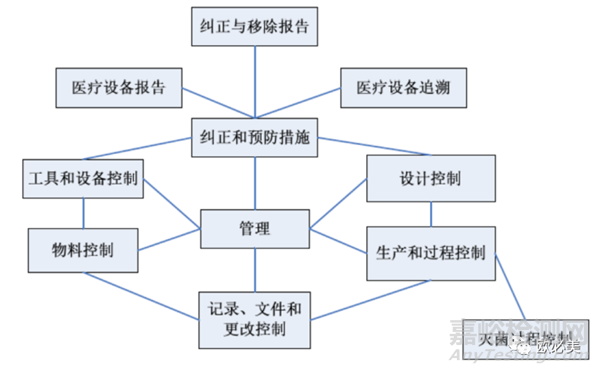
根据QSIT,事先通知的检查Pre-announced Inspections(PMA体考属于该类检查)主要针对QSR820七个子系统展开,分别是:管理控制、纠正和预防措施(CAPA)、设计控制、生产和过程控制(P&PC)、记录/文件/变更控制、材料控制、设施/设备控制。其中的管理控制、纠正和预防措施(CAPA)、设计控制、生产和过程控制(P&PC)为四个核心子系统,它们之间的关系为:
根据FDA要求,PMA体考必须包含四个核心子系统(上市后常规检查通常不会全部审),审核流程上,如QSIT,通常按照如下流程展开:
管理→设计开发→纠正和预防措施→生产和过程控制→回到管理控制。
1、管理
总体要求和上市后常规检查无区别,关注整个质量体系的策划,主要包括质量方针、质量目标、组织框架、职责和权限、管代、内审和管评这些。
虽然是上市前体考,FDA通常依旧要求提供过往数年的内审、管评记录供审核,除非企业体系新建立,或声称近期才符合QSR 820。由于PMA体考是在PMA文档(包括了质量手册、程序文件和主要SOP)提交之后开始,因此,质量体系运行的时间是需要保持描述上一致性。
2、设计开发
合理的设计开发流程策划;
设计开发输入需要识别FDA法规和标准,和已提交的PMA技术文档保持一致。考虑到大部分企业申请FDA都是产品研发后才开始,故而初始研发文件并没有识别FDA法规,另外适用标准版本通常也有更新,因此,企业需要通过设计开发变更流程纳入新的设计输入要求,作为对旧研发文档的补充;
已提交的PMA技术资料,如各类研究资料,测试报告,报告时间不能和研发线冲突;
研发可追溯性资料,企业通常需要编制追溯文件(如追溯表),将产品性能功能与设计需求、设计文档、设计验证和确认的证据进行追溯;
若企业在提交PMA技术资料和PMA现场体考之间,已经对产品或工艺进行了重大变更,则FDA审核员需要和CDRH/OC或OIVD技术评审团队讨论后再决定是否开具483(不符合项);
3、纠正和预防措施
不良事件报告、纠正和移除报告、医疗器械跟踪在CAPA系统下;
其它系统与CAPA系统的关联,什么情况下触发CAPA,比如客诉系统、不合格品控制、管理评审等;
CAPA流程的合理性和完整性,原因分析、纠正、纠正措施、有效性验证(如三个月跟踪)等;
虽然是上市前体考,FDA依然会要求提供过往数年的CAPA记录。
4、生产和过程控制
生产过程、设备与环境、检验与不合格品、采购、人员培训,都归在本系统下,因而这些资料之间的串联需要注意,比如不同资料之间的日期关联、人员关联等;
工艺确认是核心关注点,包括质量体系使用的计算机软件的确认;
灭菌过程,有独立的审核流程,具体见QSIT。
综合来讲,PMA体考属于Level 2 Comprehensive Inspections级别审核,教上市后例行审核有更多的审核内容和更严要求。另外,现场研发文档、体系资料与事先提交的PMA技术文档中体系资料的一致性非常重要,FDA现场审核员拥有这部分资料,会拿出来核对。