摘要 目的:查找注射剂和滴眼剂生产的关键控制点,制定预防措施,为注射剂和滴眼剂提供良好的质控保障。方法:调研两家医药企业,采用危害分析和关键控制点法、失败模式影响分析等风险管理工具,查找生产过程的风险点,开展风险评估和风险控制,制定预防措施。结果:对风险系数处于高风险和中等风险水平的风险源采取控制措施后,各风险点的风险等级有所降低。结论:注射剂和滴眼剂原辅包须与制剂的生产环境相匹配,生产监测的全面化与规范化有待提升。
2017年国家食品药品监督管理总局颁布《总局关于调整原料药、药用辅料和药包材审评审批事项的公告》(2017年第146号),将原料药、辅料、药包材与药品制剂进行关联审评,进一步体现原辅包在制剂研发、生产全过程中的重要价值。
对生产环境的洁净需求在医药企业的生产中占有核心的地位,其洁净效果直接影响产品的质量。随着监管理念对生产过程控制的重视及2010年版《药品生产质量管理规范》(Good Manufacturing Practice of Medical Products,GMP)的实施,我国对药品及其原辅包材料生产洁净车间环境指标的确认和验证要求越来越高。为更有效地控制洁净室的环境,需在洁净室监控中引入质量风险评估和管理的理念,利用日常监测数据进行污染与风险预测。但在实际生产中,多数原辅包生产企业对其产品的质量控制尚停留在产品检验阶段,对其产品及生产环境的监控还未引入质量风险评估和管理的理念。
质量风险管理在各行各业的设计及生产管理过程中应用广泛并取得了良好效果。在制药行业,最早由美国食品药品管理局(Food and Drug Administration,FDA)于2002年提出;2005年,人用药物注册技术要求国际协调会(ICH)发布Q9质量风险管理指南,指出应在药品的全生命周期内对药品质量风险进行评估、控制、沟通和审核;我国2010年版《药品生产质量管理规范》(GMP)引入了质量风险管理理念。已有研究将质量风险管理的方法应用在口服固体制剂[1]、注射剂[2-5]以及滴眼剂[6]等的生产管控和生产环境的风险评估中[7-9],但目前尚未见针对无菌制剂辅料和药包材生产环境的研究。
本研究以风险较高的注射剂和滴眼剂为研究对象,采用风险评估管理工具——危害分析和关键控制点(Hazard Analysis and Critical Control Points,HACCP)、失败模式影响分析(Failure Mode Effects Analysis,FMEA),对注射剂、滴眼剂及原辅包生产工艺各环节的环境进行风险识别、分析和评价,查找关键控制点,制定预防措施,为类似无菌制剂提供良好的质控保障。
1、研究方法
1.1 质量风险管理过程
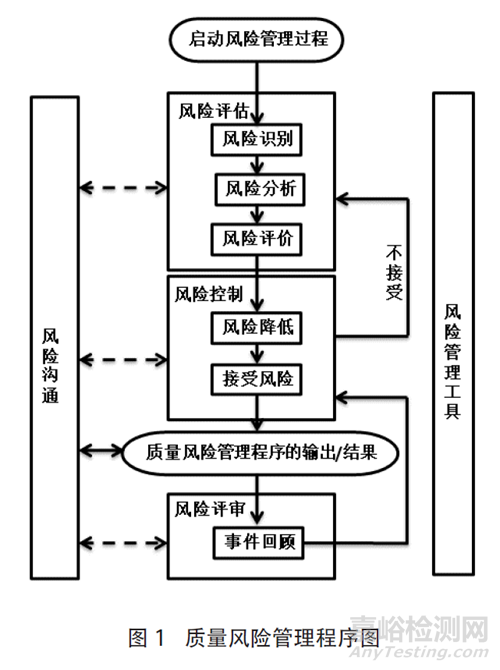
典型的质量风险管理程序见图1,风险管理程序启动后,主要分为以下几部分进行[10]。
1)风险评估 风险评估包括三方面的内容:风险识别、风险分析和风险评价。风险识别-找出容易出错的地方(风险点);风险分析-分析风险发生的危害及危害发生的几率,可进行定性或定量评估;风险评价-评价危害的严重性,得出风险级别。
2)风险控制 研究用何种方式来降低或消除风险,以及风险降低后是否达到可接受的水平。
3)风险沟通与审核 在整个风险管理流程的任意环节均可作出决定,可以返回上一步收集更多信息,对风险模型进行调整,或根据信息终止风险管理流程。
1.2 风险管理工具
本研究使用的风险管理工具有:
1)危害分析和关键控制点(HACCP)
HACCP是一种用于保证产品可靠性和安全性的预防性工具,对过程的每一步进行监视和控制,以降低危害发生的概率。
2)失败模式影响分析(FMEA)
FMEA是在产品和过程设计阶段,对各个工序进行分析,找出所有潜在失效模式,并分析其可能的后果,从而预先采取必要措施,以提高产品质量和可靠性。
应用FMEA的步骤如下:第一,对产品及其生产过程进行分析;第二,列出产品潜在的失效因素,估计失效发生的频率、严重度及可检测水平,并按照评估得分,计算风险系数(Risk Priority Number,RPN);第三,进行问题的总体评估,提出改正的措施及控制失效发生的方案。
2、研究过程
实地调研和现场监测A和B两家制药公司的生产车间洁净环境。通过座谈、问卷等方式收集生产管控相关资料,综合洁净环境调研和现场监测的内容,采用HACCP,查找注射剂、滴眼剂及其原辅包材料生产环境的风险点,进而采用FMEA进行风险评估和风险控制。
2.1 滴眼剂及其原辅包材料的生产环境调研分析
滴眼剂是由药物与适宜辅料制成的供滴入眼内的无菌液体制剂[11]。滴眼剂的制备工艺主要有三种:一是采用吹灌封一体机进行制瓶-灌药-封口过程;二是用无菌滴眼剂瓶进行无菌灌装;三是外购内包材,对其进行洗-烘-灭处理后进行无菌灌装[12]。吹灌封技术在2010年被写入我国GMP[13],目前许多滴眼剂生产企业采用该技术生产滴眼剂[14]。
内包材和注射用水是滴眼剂生产过程两个需要重点关注的风险要素。滴眼剂的内包材即滴眼剂瓶,材质多为低密度聚乙烯、聚丙烯、聚氯乙烯、聚酯类等[12],在高温下易变形,多采用无菌工艺制备[15]。配制滴眼液需使用注射用水。注射用水的制备工艺:以纯化水为原水,经多效蒸馏水机处理,形成纯化蒸汽,纯化蒸汽经冷凝成为注射用水,进入储罐供使用[16]。
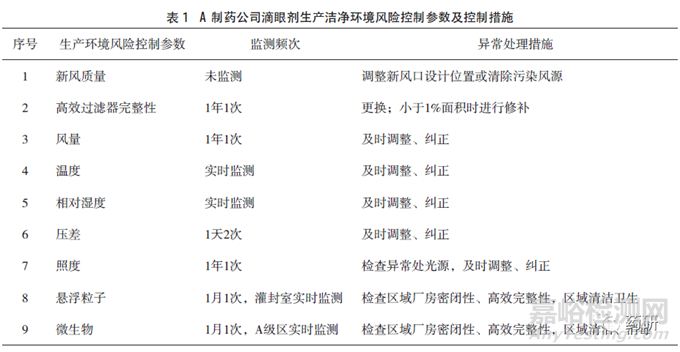
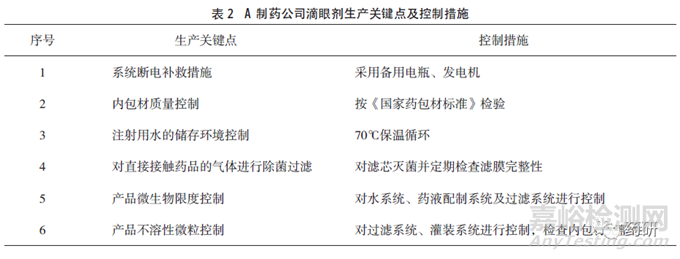
A制药公司有多条生产滴眼剂的吹灌封生产线,注册批准工艺中生产车间的洁净级别要求为C+A级,日常监测可达到规定级别,每年对洁净环境控制参数进行一次验证。通过调查获得的该企业产品生产洁净环境风险控制参数及措施见表1,生产关键点及控制措施见表2。调研过程发现生产车间内微生物监测数值较高的区域为人/物流区域及清洗区域,车间控制微生物的措施有高温高压融化粒料,除菌过滤直接接触药品的气体,蒸汽灭菌每批产品生产使用的滤芯并检查滤膜完整性。
2.2 注射剂及其原辅包材料的生产环境调研分析
注射剂指原料药物或与适宜的辅料制成的供注入体内的无菌制剂。注射剂直接入血,起效迅速,属于高风险剂型[4]。注射剂最受关注的质量问题是可见异物[2],带有可见异物的注射剂一旦被注入人体,会给患者带来难以预知的安全隐患[17-18]。注射剂作为高风险产品,生产过程控制要求高,对厂房环境和设备的运行、操作有着严格要求[19]。注射剂是典型的最终灭菌工艺生产的无菌制剂,生产中无菌保证和对可见异物的控制是较大挑战。
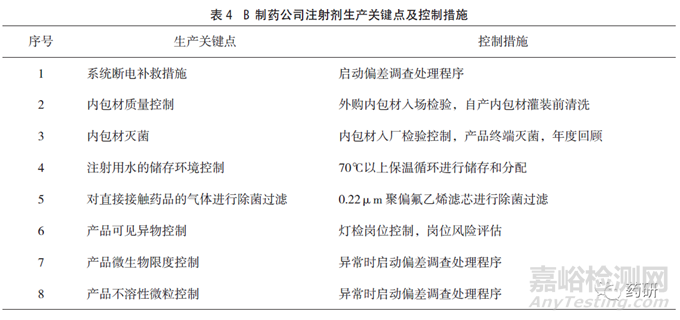
B制药公司具有多条注射液产品线,其注册批准工艺中生产车间的洁净级别要求为C+A级,日常监测可达到规定级别,对空调系统的再验证周期为2年,每年定期评估,其生产环境风险控制参数及措施见表3。车间生产线安装有粒子自动监测系统,微生物控制建立有相应标准。其他生产关键点和控制措施见表4。
2.3 注射剂、滴眼剂及其原辅包材料的生产环境风险管理
对于生产过程中关键控制点的确定,采用世界卫生组织(World Health Organization,WHO)食品法典委员会推荐的关键控制点判断树[20]。通过对注射剂、滴眼剂及其内包材和原辅料生产的工艺流程进行危害分析,结合实地检测与调研发现的问题,判断出生产过程每一步骤中存在的潜在危害,对危害的严重程度和可能性进行分析、评价,确定风险关键控制点。
接下来运用FMEA方法,对风险源的严重性、可能性和可检测性进行分析和评估,并按照评估得分,计算RPN。评定标准分别见表5、表6和表7。
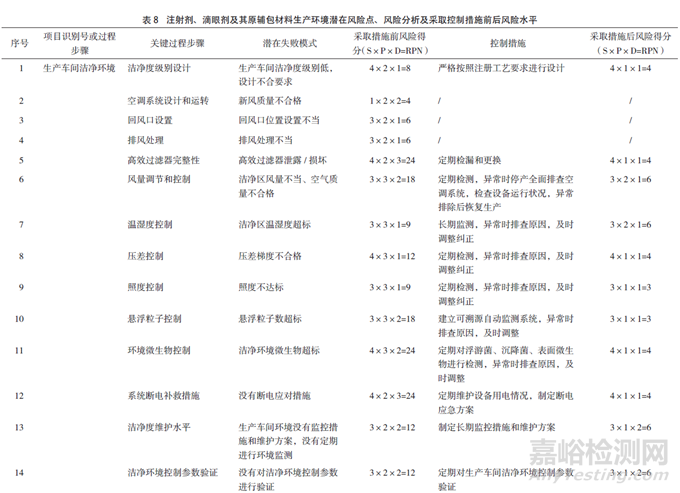
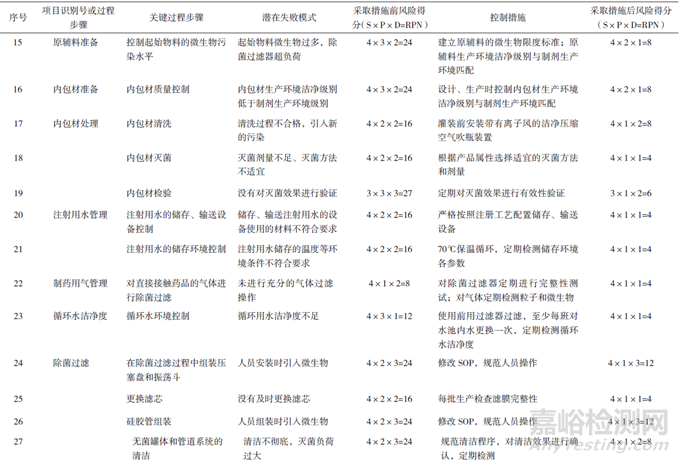
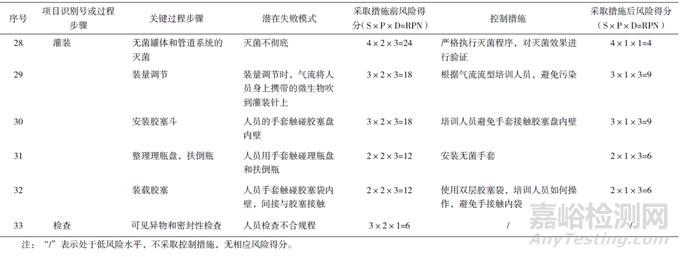
RPN为严重性(对产品质量的影响程度)、可能性(风险产生的可能性程度)和可检测性(潜在风险造成危害前,对其发生的可能性进行监测)三者的乘积。当RPN>16时,风险等级为高风险水平;当8≤RPN≤16时,为中等水平;当RPN<8时,为低风险水平。对注射剂、滴眼剂及其原辅包材料的生产环境评估结果见表8。
风险分析可知,生产洁净环境各关键过程步骤中,风险等级比较高的有高效过滤器完整性、风量调节和控制、环境微生物以及悬浮粒子的控制等。生产洁净车间若没有断电应对措施,危害较大,因为高效系统全部靠电力维持。洁净车间在设计时通常能够根据产品属性选择符合GMP要求的洁净度级别以及温湿度、照度等参数的风险等级处于中等风险水平。生产车间洁净环境控制参数的验证,以及拟定可行的监控措施和维护方案同样是比较重要的方面,若没有相应措施,生产车间洁净环境难以长期维持稳态。
在原辅包准备方面,若不控制起始原辅料和包装材料的微生物水平,或控制措施不到位,如内包材的清洗、灭菌及灭菌效果验证没有配套规定方案,会使最终产品染菌,风险较高。结合生产工艺对可能引入微生物的风险点进行分析,注射用水和制药用气的储存、运送环境都需要严格管控,除菌过滤及灌装等步骤都包含高风险操作,也需要严格控制。
接下来对风险系数处于高风险和中等风险水平的风险源拟定控制措施,并对其进行控制后的再评估,分析风险接受水平(表8)。采取控制措施后,各风险点的风险等级均有所降低,生产车间洁净环境方面,需要按照注册工艺要求进行总体设计,对洁净环境各参数定期验证,制定监控措施及预警和纠偏机制,并设立风险预案,遇到异常及时排查纠正。原辅料生产环境需要同制剂生产环境相匹配,制剂生产也需要对原辅材料及包装材料进行检验及质量控制。生产各环节尤其是容易引入微生物的风险点要格外重视,严格规范各步骤,制定监控措施和维护方案,注射剂、滴眼剂及其原辅包材料生产环境风险水平将得到降低并达到可接受的水平。
3、讨论
3.1 原辅包生产环境须与制剂的生产环境相匹配
原料药、辅料、药包材与药品制剂关联审评审批,要求原辅包的生产环境与制剂的生产环境相匹配,满足制剂的需求。目前我国还未有相关标准或规范明确规定药用辅料生产环境的洁净度,许多药用辅料生产企业尚未建立在洁净环境中生产药用辅料的理念,即使生产车间为洁净环境,也难以与药物剂型要求相匹配。例如,注射剂生产企业采购辅料时,应采购C级生产环境下生产的辅料,但市面上的辅料多在D级环境下生产。在关联审评审批的背景下,有必要建立健全针对药用原辅料生产环境洁净度要求的相应标准,督促药用原辅料生产厂家提高生产环境洁净级别,使药用原辅料生产的洁净环境与制剂生产相匹配。
3.2 生产洁净环境监测需要全面化与规范化
悬浮粒子与微生物是洁净环境的两个重要参数,风险分析中亦发现二者处于高风险等级。国内外GMP明确要求对悬浮粒子进行动态监测。对于无菌制剂生产的A级环境,悬浮粒子应设置在线实时自动监测系统,对于不同洁净环境级别的关键房间,应做定期监测。国内外GMP要求对微生物制定严格的监控方案,定期监测不同洁净级别房间的浮游菌、沉降菌、表面接触菌。调研过程中发现微生物监测数值较高的房间为人流、物流区域以及清洗区域,如洗手、更衣室等,该类房间洁净级别较低。风险分析过程显示,人员洁净服携带的微生物存在被气流吹至无菌生产线的风险,因此不能忽视洁净级别较低房间的微生物控制。另外,洁净环境悬浮粒子和微生物的监控都应当制定应急处理方案,若发现异常,及时启动偏差处理措施,排查原因后处理。
风量调节和控制对于洁净环境也是一个高风险点,洁净区风量不当或空气质量不合格容易在无菌制剂的生产过程引入微生物,严重影响产品质量。对风量应当定期监测,很多企业对于风量的监测是每季度一次,但监测频次应尽可能增加,以随时掌握风量状态,出现异常时,应立即停产对空调系统进行全面排查,检查系统设备的运行情况、过滤器是否存在破损或堵塞等,待异常排除后再恢复生产。
温湿度与压差是洁净环境比较常规的监测项目,风险分析发现,该类指标出现异常的可能性很大,生产过程(如配液时的高温高湿操作)会导致环境温湿度的波动,应设置实时监控系统进行长期监测,异常波动时由控制系统自动进行除湿降温处理。压差是保证气流从洁净等级高的区域流向洁净等级低的区域的一个关键因素,对压差梯度也应实时监测,并有相应自控系统在出现异常时进行风险提示,及时采取措施。
4、结论
在关联审评审批的背景下,药用原辅包必须在与制剂生产相匹配的环境下进行生产,生产洁净环境各参数的异常都会对产品质量及用药安全造成较大风险,需要对洁净环境各参数制定严格且正确的监测方案,以期为无菌制剂提供良好的质控保障,保障人民用药安全。